4. Tarcie ciał stałych
4.1. Tarcie zewnętrzne ciał stałych
Badanie tarcia zewnętrznego jest utrudnione ze względu na to, że jakkolwiek jest ono zjawiskiem mechanicznym, a więc łatwym do wykrycia i ilościowego zmierzenia, to jednak u jego podstaw leżą oddziaływania międzycząsteczkowe zachodzące w warstwie wierzchniej ciała stałego, zależne m.in. od składu i struktury materiałów trących. Zasięg oddziaływania tarcia w głąb materiału jest bardzo mały i dlatego istotny wpływ na wartość oporów tarcia ma stan powierzchni, jej zanieczyszczenie, stopień nawilgocenia oraz skład atmosfery, w której pracuje maszyna.4.1.1. Tarcie spoczynkowe (statyczne) ciał stałych
Ciało stale o ciężarze G spoczywa na równi pochyłej (rys. 4.1). Przemieszczaniu ciał przeciwstawia się siła tarcia T styczna do płaszczyzny ruchu, przeciwnie skierowana do kierunku ruchu i kierunku działania siły przemieszczającej ciało. Przyczyną
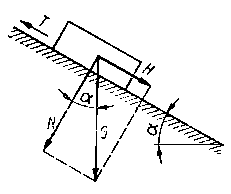 | Rys. 4.1. Układ sił działających na ciało spoczywające na równi pochyłej |
występowania siły tarcia są zjawiska występujące przy stykaniu się dwóch ciał stałych. Dla ciała znajdującego się w spoczynku na równi pochyłej (rys. 4.1), o kącie nachylenia a, można zbudować trójkąt sił G, N, T (T = H). Występują wtedy następujące zależności
| T = G sin a; N = G cos a | (4.1) |
a więc współczynnik tarcia
| µ = |
T N | = |
G sin a G cos a | = tg a | (4.2) |
| To = µs G | (4.3) |
Z rys. 4.1 wynika, że
| G ² = N ² + To² | (4.4) |
oraz
| tg ao = |
To N | (4.5) |
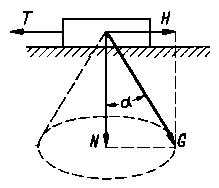 | Rys. 4.2. Stożek tarcia |
tarcia dąży do pewnej wartości skończonej. Wartość tej siły określa się mierząc najmniejszą wartość siły potrzebnej do zmiany stanu względnego spoczynku ciała i nazywa się ją siłą tarcia spoczynkowego (statycznego). Z tego wynika, że w zależności od wartości siły przyłożonej z zewnątrz siła tarcia spoczynkowego może przyjmować dowolne wartości od 0 aż do Tmax, a kierunek jej działania może być różny. Tylko graniczna wartość tej siły jest określona i równa granicznej wartości siły przyłożonej z zewnątrz, powodującej ruch ciała.
Tablica 4.1. | Skojarzenie | µs | Skojarzenie | µs | w powietrzu | w próżni | w powietrzu | w próżni | Cu-Ni | 0,45 | 1,50 | Cu-Fe | 0,51 | 0,75 | Ta-Ni | 0,23 | 0,90 | Ta-Cu | 0,44 | 0,43 | Wo-Fe | 0,19 | 0,40 | Wo-Cu | 0,34 | 0,41 | Wo-Ni | 0,21 | 1,36 | |||||||||
4.1.2. Tarcie ruchowe (kinetyczne) ciał stałych
Wartość współczynnika tarcia ruchowego jest mniejsza od wartości współczynnika tarcia spoczynkowego. Rozróżnia się dwa rodzaje tarcia ruchowego ciał stałych, a mianowicie: tarcie fizycznie suche i tarcie technicznie suche. Ten podział dotyczy wyłącznie bardzo dokładnych badań laboratoryjnych. Praktycznie istnieje prawie zawsze tarcie techniczne suche. Tarcie fizycznie suche występuje wtedy, gdy dwie współpracujące powierzchnie ciał stałych stykają się bezpośrednio, nie rozdzielone ciałem trzecim.
Tablica 4.2. | Skojarzenie | Tarcie ruchowe µr | Tarcie spoczynkowe µs | Skojarzenie | Tarcie ruchowe µr | Tarcie spoczynkowe µs | Stal-żeliwo | 0,18 | 0,30 | Stal-poliestry | 0,11 | 0,20 | Stal-stal | 0,10 | 0,17 | Stal-poliamidy | 0,05 | 0,10 | Stal-mosiądz | 0,15 | 0,19 | Stal-teflon | 0,04 | 0,09 | Żeliwo-żeliwo | 0,16 | 0,28 | Stal-AKF | 0,20 | 0,45 | Stal-żeliwo | 0,15 | 0,32 | Stal-nylon | 0,08 | 0,12 | Stal-Cu | 0,10 | 0,22 | Stal-szkło | 0,08 | 0,19 | Stal-grafit | 0,08 | 0,12 | Stal-agat | 0,19 | 0,38 | Cu-grafit | 0,10 | 0,14 | Stal-rubin | 0,11 | 0,13 | Cu-miedzio-grafit | 0,15 | 0,26 | Stal-fibra | 0,12 | 0,14 | |||||
4.1.3. Tarcie ślizgowe
4.1.4. Tarcie toczne
Tarciem tocznym nazywa się taki rodzaj tarcia, przy którym prędkości obu ciał w punktach ich wzajemnego styku są równe, a czas trwania styku tych punktów w przypadku ciał idealnie sztywnych dąży do zera. Ruch jednego ciała względem drugiego sprowadza się do obrotu ciała wokół osi przechodzącej przez punkty styku i leżącej na płaszczyźnie stycznej do obu ciał. Tarcie toczne występuje w punktach styku, w których odkształcenie sprężyste materiału powoduje styk strefowy na pewnym obszarze. W strefie styku występuje tarcie ślizgowe zewnętrzne na granicy styku oraz tarcie wewnętrzne w odkształconej objętości warstwy wierzchniej trących ciał.
Tocząca się kulka zgniata materiał, który stawia jej opór w strefie styku. Reakcja normalna, równoważąca siłę normalną obciążającą kulkę, jest przemieszczana w kierunku ruchu kulki od jej osi symetrii o pewną wartość. Siła normalna oraz reakcja normalna tworzą parę sił (moment oporu ruchu kulki). Moment oporu jest momentem tarcia tocznego. Wartość współczynnika tarcia tocznego zależy od własności sprężysto-plastycznych metali oraz od wartości siły normalnej (obciążenia). Im większa twardość stykających się ciał, tym mniejsza wartość współczynnika tarcia tocznego (wypadkowe z nacisków tworzą ramię/). Moment tarcia jest równoważony przez moment, jaki tworzy siła tarcia (styczna) pomnożona przez średnicę toczącej się kulki.
Mt = N f = N µobl d
(4.6)
gdzie µobl - obliczeniowy współczynnik tarcia tocznego µobl/d = µ/d.
Na podstawie podanej zależności można stwierdzić, że kulka może się toczyć tylko wtedy, jeśli siła popychająca P jest równa lub większa od siły tarcia T
P ≥ T ≥ N µobl
(4.7)
gdzie: T - siła tarcia w obszarze styku elementów tocznych, N - nacisk normalny kulki, µobl obliczeniowy współczynnik tarcia (p. wzór (4.6)).
Wartość obliczeniowego współczynnika tarcia tocznego µobl w zastosowaniach technicznych wynosi 0,05-0,0005. Na przykład wartości współczynnika tarcia tocznego w łożyskach tocznych (określonego jako stosunek siły popychającej łożysko P do obciążenia normalnego N) podano w tabl. 4.3.
Tablica 4.3.
Wartości współczynnika tarcia łożysk tocznych
Typ łożyska Współczynnik tarcia
µr
Kulkowe zwykle przy obciążeniu promieniowym 0,002
Kulkowe zwykłe przy obciążeniu osiowym 0,004
Kulkowe wahliwe 0,001 5
Walcowe z wałeczkami walcowymi krótkimi 0,003
Walcowe z wałeczkami walcowymi długimi 0,006
Igiełkowe 0,008
Baryłkowe 0,004
Kulkowe wzdłużne 0,003
Stożkowe przy obciążeniu promieniowym 0,008
Stożkowe przy obciążeniu osiowym 0,020
Współczynnik tarcia tocznego łożysk zależy nie tylko od obciążeń normalnych, ale także od obciążeń osiowych, wywołujących częściowo tarcie między kulkami a bocznymi powierzchniami bieżni. Ze wzrostem prędkości względnej toczenia kulek wartość współczynnika tarcia µobl maleje.
4.1.5. Hipotezy tarcia suchego ciał stałych
Mimo długotrwałych badań występującego powszechnie w przyrodzie i technice zjawiska tarcia zewnętrznego ciał stałych, nieodłącznie związanego z wszelkiego rodzaju ruchem w ośrodku materialnym, nie powstała dotychczas ogólna teoria tarcia suchego.
Ogólna teoria tarcia suchego powinna zawierać zależność umożliwiającą określenie wartości współczynnika tarcia, potwierdzalną eksperymentalnie w dowolnych warunkach badań. Tarcie suche jest procesem złożonym, niejednorodnym,
Tablica 4.4.
Ważniejsze hipotezy tarcia suchego
Grupy teorii Nazwa teorii Podstawowe wzory Cechy teorii
Mechaniczne Amontonsa
Coulomba T = µ NT = A + µ N
Teoria pokonywania nierówności powierzchni; nie zależy od N, A nie zależy od rzeczywistego pola styku
Bowdena T = S tpl + Sw pw Teoria tworzenia i niszczenia szczepień; opory tarcia są sumą oporów ścinania nierówności szczepionych i przepychania odkształconego materiału
Molekularne Tomlinsona µ = kE
npx
Opory tarcia są oporami pokonywania przyciągania molekularnego
Dieriagina T = µ(N+No)
N = PoSr Pokonywanie chropowatości molekularnej ; nie uwzględnia własności materiału
Molekularno-mechaniczna Kragielskiego µ = T
N = aS
N +a
Tarcie = suma oporów pokonywania szczepień, chropowatości i przyciągania molekularnego; a= f(ścinania); b = f( powiększania oporu na ścinanie od N).
T - siła tarcia,
N - siła skierowana prostopadle do powierzchni tarcia,
µ - współczynnik tarcia,
A - siła adhezji,
tpt - wytrzymałość na ścinanie połączeń tarciowych,
S - pole rzutu powierzchni styku,
Sw - pole przekroju poprzecznego bruzdy wyciskowej w procesie tarcia,
Sr - pole powierzchni rzeczywistego styku,
Pw - średni jednostkowy opór wyciskania materiału,
Po - średni jednostkowy opór przyciągania molekularnego,
E - energia zużywana na rozerwanie pary zetkniętych cząstek,
n - liczba par zetkniętych cząstek,
p - średnia wartość siły odpychania cząstek,
x - droga jednostkowa przemieszczających się cząstek
l - odległość pomiędzy środkami kulistych cząstek,
k= x
l
a - współczynnik zależny od adhezyjnych własności ciał trących,
b - współczynnik zależny od mechanicznych własności ciał trących.
określonym przez zespół różnych zjawisk zależnych od parametrów i warunków tarcia. Począwszy od XV wieku (Leonardo da Vinci) wielu wybitnych uczonych przeprowadzało badania zagadnień tarcia i formułowało zależności, z których można obliczyć wartość współczynnika tarcia.
Udoskonalane metody badań i aparatura pomiarowa umożliwiają bardziej dogłębne i szczegółowsze poznanie natury tarcia. Opracowane na tej podstawie współzależności są słuszne w pewnym zakresie warunków tarcia. W ten sposób powstało wiele hipotez i teorii tarcia suchego (tabl. 4.4).
4.1.5.1. Mechaniczne teorie tarcia suchego
Mechaniczne teorie tarcia suchego były najwcześniej opracowane. Teorie te objaśniają opór tarcia jako czynnik przeciwdziałający wykonywanej pracy, zużywanej na: unoszenie ślizgającego się elementu maszyny po nierównościach powierzchni drugiego elementu, na ścinanie nierówności i połączeń tarciowych oraz na pokonanie oporów wywołanych odkształceniami sprężystymi i plastycznymi w mikroobszarach styku.
Teorie te rozpatrujące zjawiska występujące w połączeniach tarciowych, których źródłem jest międzycząsteczkowe współoddziaływanie trących powierzchni, nie uzależniają oporów tarcia od przyczyn adhezyjnego sczepiania, lecz interpretują je z mechaniczno-wytrzymałościowego punktu widzenia.
Mechaniczna teoria Leonarda da Vinci. Teoria ta opierała się na spostrzeżeniach, że „zdolność ciał do ślizgania się jest różna i dlatego tarcie ma różną wartość”. Ciało o bardziej gładkiej powierzchni ma mniejszą wartość tarcia niż ciało chropowate. Dowolne ciało wykazuje przy tarciu opór równy około jednej czwartej swojego ciężaru pod warunkiem, że styka się wyrównana płaszczyzna z polerowaną płaszczyzną. Badając tarcie liny zwiniętej w zwój i rozciąganej Leonardo da Vinci wyciągnął wniosek, że siła tarcia jest niezależna od powierzchni styku.
Teoria Amontonsa. W końcu XVII w. M. Amontons sformułował prawo tarcia suchego. Stwierdził on mianowicie, że tarcie jest rezultatem wspinania się jednego ciała po nierównościach drugiego przy ich przesuwaniu względem siebie pod działaniem nacisku normalnego. Prawo tarcia suchego wyraził on w postaci ogólnie znanego wzoru Amontonsa
T = µ N
(4.8)
Wzór Amontonsa przetrwał do tej pory i jest powszechnie używany w technicznych obliczeniach, pomimo że jest on mało dokładny. Ze wzoru tego wynika, że współczynnik tarcia µ nie zależy od obciążenia normalnego N. W rzeczywistości, jak to wykazał w swoich doświadczeniach m.in. Dieriagin, współczynnik tarcia zależy od obciążenia, a oprócz tego od mechanicznych, geometrycznych i chemicznych właściwości powierzchni trących. Współczynnik tarcia statycznego rośnie ze wzrostem czasu styku powierzchni tarcia ciał stałych.
Wzrost wartości współczynnika tarcia jest szczególnie duży przy kojarzeniu materiałów plastycznych i półplastycznych. Pod wpływem obciążenia zewnętrznego występują wtedy odkształcenia plastyczne stykających się nierówności (rys. 4.3), a zatem rośnie powierzchnia rzeczywistego styku i siły adhezji zwiększają wartość współczynnika tarcia, czego nie uwzględnia wzór Amontonsa.
Należy zwrócić uwagę na to, że wg Amontonsa siła tarcia nie zależy od nominalnej powierzchni styku z wyjątkiem przypadku, gdy ze zmianą pola powierzchni styku zmienia się nacisk jednostkowy. To istotne ograniczenie, wynikające z prawa Amontonsa, często jest przez wielu autorów pomijane.
Rys. 4.3. Model oddziaływania obszarów styku trących powierzchni
Amontons stwierdził, że „siła tarcia znajduje się w złożonej zależności od nacisku jednostkowego, czasu i prędkości ślizgania przy tarciu ruchowym". Prawo Amontonsa zakłada, że trące ciała są idealnie sztywne.
Coulomb w końcu XVIII w. sformułował prawo tarcia suchego i wyraził je za pomocą następującej zależności
T = µ N + A
(4.9)
W równości tej składnik A jest poprawką do wzoru Amontonsa i przedstawia część siły tarcia zależnej od molekularnego oddziaływania (sczepności) powierzchni trących.
Coulomb przyjął jednak wartość składnika A jako stałą dla płaskich powierzchni, nie uzależniając jej od wartości nacisku normalnego ani od jakości powierzchni styku.
Teoria Bowdena. Teoria ta jest oparta na hipotezie uwzględniającej charakter plastyczny odkształceń rzeczywistej powierzchni styku trących ciał stałych. Stanowi ona uogólnienie badań eksperymentalnych kojarzonych metali. Bowden prowadził badania na stanowisku zwanym przyrządem Bowdena-Lebena. Na podstawie wyników badań wysunął hipotezę, że wartość współczynnika tarcia oraz charakter uszkodzeń powierzchniowych, powstałych przy tarciu ślizgowym są określone głównie względnymi wartościami własności fizycznych trących się powierzchni ciał stałych. Charakter współoddziaływania trących powierzchni zależy od wartości stosunku ich twardości, a przy dużych wartościach pracy tarcia również od temperatury obszarów styku (będącej funkcją obciążenia i twardości materiału) oraz od temperatury topnienia trących materiałów.
Przy takich założeniach Bowden podzielił wszystkie przypadki tarcia ślizgowego na trzy rodzaje:
- ślizganie się metalu twardego po metalu twardym,
- ślizganie się metalu miękkiego po metalu twardym,
- ślizganie się metali o jednakowej twardości.
Procesy tarcia metali nie mogą być uważane wyłącznie za zjawiska powierzchniowe. Podczas tarcia suchego następuje tworzenie, a następnie ścinanie metalicznych połączeń współpracujących elementów.
Od rodzaju styku trących powierzchni, określonego ich chropowatością, własnościami mechanicznymi materiałów i wartością obciążenia normalnego przypadającego na jednostkę powierzchni nominalnej, uzależnione są odpowiednie wartości rzeczywistych nacisków jednostkowych. W obszarach styku działają siły adhezji powodujące tworzenie połączeń tarciowych trących metali, charakterem zbliżonych do połączeń powstających przy spajaniu na zimno. Podczas tarcia połączenia te są ścinane. Nie ma ujednoliconego poglądu na to, jakie własności ciał przyczyniają. się do powstawania połączeń tarciowych, jaki jest mechanizm powstawania tych połączeń, jakie są ich własności oraz jaki mają wpływ na procesy tarcia i zużywania.
Adhezyjne sczepianie jest spowodowane plastycznym płynięciem materiału w obszarach styku. Zespół zjawisk w czasie styku trących powierzchni powoduje niszczenie warstewek adsorpcyjnych lub tlenkowych na tych powierzchniach, co umożliwia utworzenie połączeń metalicznych.
Praca tarcia, zamieniana w ciepło, powoduje wzrost temperatury w obszarach styku, a przez to obniżenie twardości materiału, wskutek czego rośnie obszar styku plastycznego. Następuje wtedy sczepianie trących metali, jako bezdyfuzyjne połączenie stykających się obszarów, wskutek wiązania metalicznego w pierwotnej granicy ich rozdziału.
Sczepianie między powierzchniami dwóch metali powstaje przy czystym metalicznie styku, po pokonaniu fizycznej bariery wiązania metalicznego. Mogą również wystąpić, jak już wspomniano, połączenia metaliczne współtrących powierzchni elementów maszyn spowodowane zgrzewaniem lokalnych obszarów styku pod działaniem wysokiej temperatury, powstałej na skutek pracy tarcia, lub w wyniku adhezyjnego sczepienia (rys. 4.4).
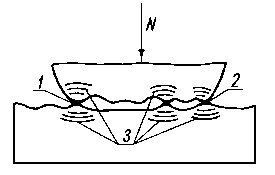
Rys. 4.4. Sczepianie się obszarów styku trących powierzchni;
1, 2 - strefy sczepienia,
3 - strefy naprężenia wokół sczepień
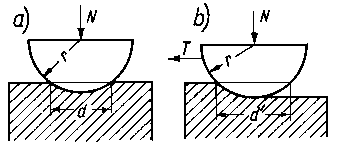
Rys. 4.5. Model Bowdena do obliczania i interpretacji siły tarcia suchego
Przy tarciu metali znacznie różniących się twardością występujące w metalu twardszym chropowatości żłobią bruzdy w metalu miększym. Wokół wyżłobionych bruzd metal jest intensywnie odkształcony plastycznie. Powstaje zatem nierównomierne umocnienie warstwy wierzchniej powierzchni trących.
Przy ślizganiu twardego ciała o kształcie półkulistym po powierzchni płaskiej metalu o znacznie mniejszej twardości i obciążonego siłą normalną N (rys. 4.5) na początku procesu następuje zagłębienie ruchomej próbki w materiał przeciwpróbki (rys. 4.5a) i zwiększenie powierzchni rzeczywistego styku. Jeżeli powierzchnia rzeczywistego styku jest tak duża, że przy danym obciążeniu odkształcenie plastyczne już nie następuje, wtedy pole rzutu powierzchni styku S na płaszczyznę jest wyrażone za pomocą następującej zależności
S =
N
Re
(4.10)
gdzie Re - granica plastyczności materiału o mniejszej twardości.
Dla uproszczenia Bowden przyjmuje, że powierzchnia styku jest idealnie gładka, a rzeczywista powierzchnia styku jest równa nominalnej powierzchni styku. Takie założenie jest uzasadnione, ponieważ chropowata powierzchnia ruchomej próbki ma zakończenie występów również w kształcie półkulistym (rys. 4.5b).
Siła tarcia wg Bowdena składa się z siły Pś niezbędnej do ścięcia powstających metalicznych połączeń i siły Pw, niezbędnej do wyciśnięcia bruzdy w miększym metalu
T = Pś + Pw
(4.11)
Siła ścinania metalicznych połączeń
Pś = Sw tpt
(4.12)
gdzie: tpt - wytrzymałość na ścinanie metalicznych połączeń w kierunku stycznym do płaszczyzny tarcia, S - jak we wzorze (4.10).
Siła wyciskania bruzdy w miększym metalu
Pw = Sw pw
(4.13)
gdzie: Sw - przekrój poprzeczny bruzdy tarcia, pw - średni jednostkowy opór wywołany wyciskaniem metalu (wartość tego oporu pw jest tego samego rzędu co granica plastyczności metalu mniej twardego).
Przekrój poprzeczny bruzdy tarcia może być wyrażony w przybliżeniu zależnością
Sw =
d ³
12r
(4.14)
gdzie: d - szerokość bruzdy tarcia, r - promień czaszy ślizgacza.
Siłę tarcia można więc obliczyć z zależności
T = S tpt + Swpw
(4.15)
Analogiczne rozumowanie umożliwiło Bowdenowi opracowanie zależności, z której można obliczyć wartość siły tarcia w przypadku, gdy kształt próbki jest cylindryczny. Parametrami jej są: promień r i długość l. Wtedy siła tarcia jest wyrażona zależnością
T = l d tpt +
d ³
12r
pw
(4.16)
Podczas badania tarcia stalowego ostrza o płaszczyznę indu przy małych prędkościach ruchu, Bowden mierzył wartość T i d. Następnie dla półokrągłego ostrza wyznaczył siłę tarcia z zależności
T =
d ³
12r
pw
(4.17)
Średnia wartość pw = 15 MPa (w 1500 G/mm2).
Granica plastyczności indu wyznaczona metodą statycznej próby wciskania kulki wynosi 10 MPa (» 1000 G/mm²).
Większa wartość oporu pw uzyskana z pomiaru tarcia jest spowodowana większą prędkością odkształcenia przy wciskaniu metalu przez przemieszczające się ślizgające ciało niż przy statycznej próbie wciskania kulki. Z badań Bowdena wynika, że opór pw nie jest równy granicy plastyczności, ale jest do niej zbliżony i od niej zależny.
Szerokość bruzdy tarcia jest zależna od granicy plastyczności, wskutek czego siła Pw niezbędna do wyciśnięcia bruzdy w miększym metalu powinna być także funkcją granicy plastyczności.
Z tego wynika, że siła potrzebna do wyciśnięcia miększego metalu przy tarciu jest tym mniejsza, im metal wyciskający jest twardszy. Siła ta dla takiej samej wartości obciążenia przy styku płaskiej powierzchni z powierzchnią kulistą jest tym mniejsza, im większa jest ilość punktów styku.
Tablica 4.5.
Wytrzymałość na ścinanie metalicznych połączeń 1) tarciowych
Metal Doświadczalne wartości wytrzymałości na ścinanie metalicznych połączeń tpt
MPa Wytrzymałość na ścinanie metalu przy próbie statycznej t
MPa
Ind 3,185 2,156
Ołów 15,680 7,350
Miedź 27,400 156,800
Stal l 372,000 882,000
1)Przy tarciu stalowego suwaka po podanych metalach przy malej prędkości ruchu.
Siła Pś ścinająca połączenia metaliczne podczas tarcia przyjmie wartość zbliżoną do wartości wytrzymałości na ścinanie miększego metalu. Podane wywody zilustrowano wynikami badań podanymi w tabl. 4.5. Analizując te wyniki różnice między wytrzymałością na ścinanie metalicznych połączeń a wytrzymałością na ścinanie samego metalu wynikają stąd, że ścinanie metalicznych połączeń następuje w głębi warstwy wierzchniej metalu o mniejszej twardości niż w obszarze umocnienia. Dowodem uzasadniającym podaną interpretację wyników badań jest pozostałość cząstek tego metalu po tarciu na stalowym ślizgaczu.
Podane w tabl. 4.5 wartości wytrzymałości na ścinanie metalicznych połączeń indu i ołowiu są rzeczywistą wytrzymałością na ścinanie metalicznych połączeń, ponieważ mała twardość tych metali zapewnia całkowity styk skojarzenia, zgodnie z założeniem podanym przy obliczaniu pola rzutu powierzchni styku na płaszczyznę. Natomiast wytrzymałość na ścinanie metalicznych połączeń miedzi i stali, podana w tabl. 4.5, jest nieco zmniejszona, ponieważ pole rzeczywistej powierzchni styku na występach nierówności jest nieco mniejsze niż pole nominalnej powierzchni styku.
Podane wyniki potwierdzają prawo Amontonsa w takim zakresie badań tarcia, w jakim pole rzeczywistej powierzchni styku zmienia się proporcjonalnie do wartości obciążenia normalnego.
Dla małych wartości siły wyciskającej, czyli dla twardych metali
T = S tpt
(4.18)
gdzie: S-pole rzutu powierzchni styku na płaszczyznę, tpt - wytrzymałość na ścinanie metalicznych połączeń.
Na podstawie poprzednio podanego modelu można wywnioskować, że dla chropowatej powierzchni pole rzeczywistej powierzchni styku zależy od wartości nacisku normalnego i granicy plastyczności metalu. Natomiast prawie nie zależy od nominalnej powierzchni styku, co jest zgodne z hipotezą Amontonsa. Na tej podstawie można sądzić, że współczynnik tarcia zależy jedynie od własności wytrzymałościowych miększego metalu rozpatrywanego trącego skojarzenia.
Ostateczny wniosek, oparty na założeniu, że ścinanie metalicznych połączeń następuje wewnątrz warstwy wierzchniej, można sformułować następująco: współczynnik tarcia jest ilorazem wytrzymałości na ścinanie miększego metalu i granicy plastyczności tego metalu. Z tego Bowden wyciąga wniosek, że wartość współczynnika tarcia dla danego metalu jest wartością stałą. Przy kojarzeniu różnych metali iloraz ten zmienia się w wąskim przedziale, dlatego przyjmuje on, że wartość współczynnika tarcia dla różnych metali jest tego samego rzędu.
Ze wzrostem temperatury wytrzymałość na ścinanie i granica plastyczności badanego metalu powinny się zmieniać proporcjonalnie. Dlatego dla powierzchni nie smarowanych wpływ temperatury na wartość współczynnika tarcia jest nieznaczny. Prawo Amontonsa przestaje obowiązywać wtedy, gdy rzeczywista powierzchnia styku nie rośnie proporcjonalnie do obciążenia.
Teoria Bowdena nie uwzględnia ani molekularnego oddziaływania powierzchni, ani wpływu chropowatości, a przyjmuje jedynie czysto mechaniczny model tarcia, uwzględniający fizyczne własności materiałów trących.
Teoria Epifanowa. Teorię Bowdena rozwinął Epifanow, który założył, że ścięcie metalu przy przesuwaniu jednej powierzchni po drugiej następuje nie tylko w punktach rzeczywistego styku, lecz także na powierzchni So kilkakrotnie przekraczającej sumaryczną powierzchnię rzeczywistego styku. Przed przednią częścią suwaka tworzy się narost metalu, a ślizganie zachodzi nie w rzeczywistej płaszczyźnie styku suwaka i próbki płaskiej, lecz wewnątrz próbki w płaszczyźnie C-C (rys. 4.6 i 4.7). Wielkość rzeczywistej powierzchni ścięcia można określić, badając szerokość i głębokość bruzdy powstałej w wyniku tarcia.
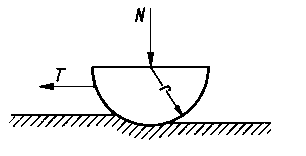
Rys. 4.6. Model Epifanowa do obliczania składowej siły tarcia suchego
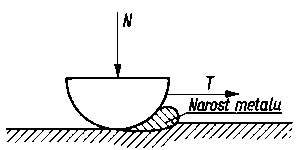
Rys. 4.7. Zagłębianie i przesuwanie narostu metalu podczas ruchu elementu
Określając rzeczywistą płaszczyznę przesunięcia przy ślizganiu i wyznaczając przy tym doświadczalne wartości siły tarcia, zmierzono wytrzymałość badanych metali na ścinanie. Wyniki były zgodne z danymi wytrzymałości metalu na ścinanie wyznaczonymi w toku badań mechanicznych metalu.
Prawo tarcia jest określone następującą zależnością
T = t Sc
(4.19)
gdzie: T - siła tarcia, t - wytrzymałość na ścinanie, Sc - powierzchnia ścinania.
Wytrzymałość na ścinanie t zależy od naprężeń normalnych sp działających w płaszczyźnie przesunięcia. Według Bridgmana
t = to + k sp
(4.20)
gdzie: to - wytrzymałość na ścinanie przy sp = 0, sp - naprężenie normalne w płaszczyźnie ścinania, k = (d t/d sp ) - szybkość zmiany wytrzymałości na ścinanie przy zmianie naprężeń normalnych (wg Bridgmana k = 0,01 ¸ 0,1).
Siłę tarcia można więc określić zależnością
T = to Sc + k Sc sp
(4.21)
a przyjmując
sp =
N
Sc
(4.22)
otrzymamy
T = to Sc + k Sc sp
(4.23)
gdzie N - obciążenie normalne.
Pierwszy człon tej zależności wyraża zmianę siły tarcia T w wyniku zmiany wartości powierzchni ścinania Sc przy założeniu, że wytrzymałość materiału na ścinanie się nie zmienia.
Drugi człon tej zależności wyraża zmianę wytrzymałości na ścinanie przy zmianie obciążenia normalnego N (w tym również udział sił adhezji).
Według Epifanowa przy wzroście obciążenia N (patrz wzór (4.23)) siła tarcia wzrasta początkowo wskutek wzrostu powierzchni ścięcia przy niezmiennej wytrzymałości na ścinanie. Jeżeli jedno z trących ciał ma dużą plastyczność, to wzrost S będzie proporcjonalny do przyłożonego obciążenia N, czyli S = N/spl
T = N
(
to
spl
k
)
(4.23)
gdzie spl - granica plastyczności metalu.
Współczynnik tarcia µ wg Epifanowa nie zależy od obciążenia normalnego N, co wynika ze wzoru
µ = to
spl k
Podane zależności zostały wyprowadzone na podstawie badań tarcia półkulistej próbki o powierzchnię płaską. Próbka była wykonana ze stali hartowanej szybkotnącej, a płaskie przeciwpróbki z miedzi elektrolitycznej, aluminium, żelaza armco, mosiądzu i innych stopów (tabl. 4.6).
W celu wykonania czystych powierzchni trących, ślizgacz poruszał się bezpośrednio za nożem skrawającym cienki wiór z powierzchni miękkich metali.
Tablica 4.6.
Wytrzymałość na ścinanie zgrzanych obszarów tarcia (uzyskana z badań tarcia i z prób statycznych)1)
Metoda określenia wytrzymatości na ścięcie Aluminium Miedź Żelazo Stal 417 Stop 437 Stop WT-2 Mosiądz
Badanie tarcia 8,3 29,2 - - - - -
7,4 24,5 56 88 106 103 42
Próby statyczne 8,0 26,5 51 81 108 92 40
W pierwszej rubryce poziomej są podane wartości obliczone z pól śladów ścięcia, w następnej obliczone z pola koła o średnicy równej szerokości bruzdy tarcia.
Wartość obciążenia zmieniano stopniowo w granicach 125 ¸ 1000 N. Siłę tarcia rejestrowano za pomocą specjalnego dynamometru. Badana siła, potrzebna do oderwania ślizgacza od powierzchni i warunkująca wytrzymałość na rozerwanie połączeń między trącymi powierzchniami, była siedmiokrotnie mniejsza od siły tarcia.
Na tej podstawie wysunięto wniosek, że powierzchnia ścinania przy tarciu jest o dwa rzędy większa niż sumaryczna powierzchnia połączeń metalicznych. Mikroskopowe obserwacje bruzd tarcia wykazały, że przed przednią częścią ślizgacza tworzy się narośl, który przemieszcza się wraz ze ślizgaczem jako jedna całość (rys. 4.8).
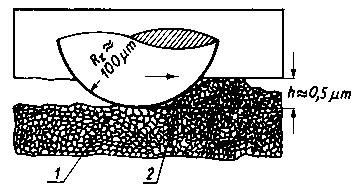
Rys. 4.8. Narost tworzący się przed czołową płaszczyzną ślizgacza;
1 - materiał odkształcony,
2 - materiał spęczony
Wzrost siły tarcia występuje dopóty, dopóki rośnie rzeczywista powierzchnia styku, przy wzroście obciążenia jednostkowego.
4.1.5.2. Adhezyjne teorie tarcia suchego
Osiągnięcia technologiczne uzyskane w obróbce powierzchniowej, dzięki której uzyskano znaczną gładkość powierzchni, nie doprowadziły do wyeliminowania przy ślizganiu dużych oporów tarcia. Omówione więc dotychczas mechaniczne teorie tarcia suchego nie wyjaśniły przyczyny tego zjawiska. Wysunięto więc hipotezę, że opór tarcia jest spowodowany oddziaływaniem pól sił atomów, cząsteczek lub jonów, a więc adhezją.
Teoria Tomlinsona. Tomlinson podał teorię, w której przyjął, że tarcie jest rezultatem adhezyjnego oddziaływania powierzchni trących ciał. Przy przemieszczaniu jednego ciała stałego po drugim stykające się powierzchnie tarcia są chropowate, a zatem poszczególne punkty materialne tych powierzchni będą od siebie oddalone różnie, a tylko niektóre będą się bezpośrednio stykały, czyli jedne cząsteczki ciał będą się przyciągały, a inne odpychały. Tylko w pewnych przypadkach siły przyciągające wystąpią jako pierwszoplanowe i między ciałami zachodzi ,,sklejenie”. Tomlinson uważa, że suma sił przyciągających jest mała i można ją pominąć. Obciążenie normalne N jest równoważone siłami odpychania (rys. 4.9 i 4.10)
N =
S
Pi
gdzie Pi - jednostkowa siła odpychająca między cząsteczkami.
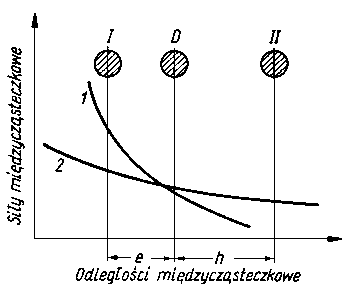
Rys. 4.9. Model Tomlinsona działania sił rniędzycząsteczkowych
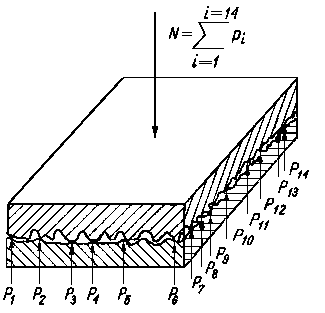
Rys. 4.10. Model Tomlinsona równoważenia obciążenia normalnego siłami odpychającymi
Podczas ślizgania po sobie dwóch powierzchni zachodzi ciągła zamiana par cząsteczek znajdujących się w zbliżeniu i równoważących obciążenie normalne. Przy przesuwaniu jednego ciała względem drugiego siły odpychania przechodzą w siły przyciągania, a te ostatnie przeciwstawiają się ślizganiu.
Przy ślizganiu jedne cząsteczki odrywają się od drugich, wychodzą ze strefy odpychania, a inne wstępują w te strefy. Ta ciągła zmiana par cząsteczek i tworzenie się nowych wiązań molekularnych powoduje rozproszenie energii E.
Jeśli przez n oznaczy się liczbę par cząsteczek znajdujących się w zetknięciu, a przez P średnią wartość siły odpychania odpowiadającej takiej parze, to
N = n P
(4.25)
Jeżeli jest znana odległość / pomiędzy środkami poszczególnych par kulistych cząsteczek, to liczba stykających się par cząsteczek przy ich przemieszczaniu na jednostkę drogi x wynosi
k = n
x
l
(4.26)
Ponieważ liczba stykających się par cząsteczek zależy od kierunku ruchu, w związku z tym Tomlinson wprowadza współczynnik poprawkowy b. Zatem
k = n b
x
l
Oznaczając średnią energię zużywaną na rozerwanie pary cząsteczek przez E, można obliczyć pracę przesunięcia LT na drodze x, która będzie równa LT = k E. Praca ta jest zużyta na pokonanie oporów tarcia. Przyjmując, że dla każdej pary cząsteczek jest słuszne prawo Amontonsa, praca tarcia
LT = µ n P x
(4.27)
a ponieważ
k E = LT
(4.28)
zatem
µ =
k E
n p x
=
E
l P
(4.29)
Wartość współczynnika tarcia jest wprost proporcjonalna do iloczynu siły odpychania i odległości poszczególnych par cząsteczek.
Liczba par cząsteczek n znajdujących się w styku zależy od wielkości rzeczywistej powierzchni styku
n = f( Sr )
(4.30)
gdzie Sr - rzeczywista powierzchnia styku zależna od obciążenia normalnego N.
Z wzorów (4.25) i (4.29) wynika, że
µ =
E b
l N
f( Sr )
(4.31)
Zatem /(jest wprost proporcjonalne do rzeczywistej powierzchni styku trących się ciał stałych.
Na podstawie badań Tomlinson ustalił zależność empirycznego obliczenia współczynnika tarcia
µ = 0,18 • 10 8 ( Ak + Ap ) 2/3
(4.32)
gdzie Ak i Ap - moduły zależne od własności mechanicznych materiału.
Thomlinson opierając się na badaniach eksperymentalnych, przeprowadzonych na znacznej liczbie próbek wykonanych z różnych materiałów, doszedł do wniosku, że ustalony przez niego współczynnik 0,18 • 10 8 ma charakter ogólny i w związku z tym wzór (4.32) określa zależność współczynnika tarcia od własności mechanicznych trących się materiałów.
Wywody Thomlinsona przeczą prawu Amontonsa-Coulomba, które wyraża niezmienność współczynnika tarcia przy zmianie nacisku jednostkowego.
Teoria Deriagina. Deriagin oparł swoją teorię na założeniu, że tarcie jest związane z molekularną chropowatością ciała, zależną od struktury materiału. Forma i wymiary atomów podczas przesuwania jednego ciała po drugim nie zmieniają się i wobec tego proces tarcia można przedstawić jako pokonywanie chropowatości molekularnej.
Na rys. 4.11 przedstawiono idealne rozłożenie atomów w płaszczyźnie styku dwóch ciał. Ciało górne pod działaniem obciążenia zajmuje ustalone położenie, przy którym jego środek ciężkości jest położony najbliżej punktu A (rys. 4.12).
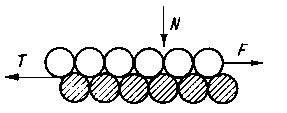
Rys. 4.11. Model mechaniczny Deriagina oddziaływania atomów w obszarze tarcia
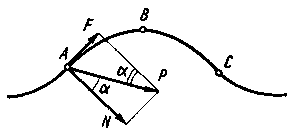
Rys. 4.12. Rozkład sił wg Deriagina podczas tarcia w obszarze styku atomów
Pod wpływem siły F górne ciało przenosi się w nowe położenie, a jego środek ciężkości podczas pokonywania chropowatości molekularnej opisze krzywa ABC (rys. 4.12). Z warunku równowagi w dowolnym punkcie krzywej, np. A otrzymamy
F
N
= tg a
(4.33)
Jeśli F/N = tg amax, gdzie amax jest maksymalnym kątem pochylenia stycznej do linii ABC, to
F = N tg amax = µ N
(4.34)
gdzie µ = tg amax - statyczny współczynnik tarcia.
Jeśli F/N = tg amax, wówczas równowaga będzie zachwiana i rozpocznie się nieprzerwane ślizganie górnego ciała po dolnym. Jeśli uwzględni się siły adhezji, wtedy prawo tarcia jest ujęte wzorem
T = (N + No) µ
(4.35)
gdzie No - siły przyciągania molekularnego.
Obliczeniowy współczynnik tarcia zmienny ze zmianą wartości N
µo =
T
N
= µ
(
1 +
No
N
)
(4.36)
gdzie µ - rzeczywisty stały współczynnik tarcia.
Siły przyciągania molekularnego No mogą mieć wartość rzędu N przy małym obciążeniu. W przypadku dużych obciążeń, a więc i przy dużych rzeczywistych powierzchniach styku wartość No rośnie proporcjonalnie ze wzrostem wartości rzeczywistej powierzchni styku.
Przyjmując No = po Sr można wtedy wzór na siłę tarcia przedstawić w następującej postaci
T = µ (N + po Sr )
(4.37)
gdzie: po - jednostkowa siła przyciągania molekularnego. Sr - rzeczywista powierzchni styku zmienna wraz ze zmianami obciążenia N.
Dla metali plastycznych można przyjąć, że
Sr =
N
p 'o
(4.38)
gdzie p 'o - wartość siły jednostkowej, poniżej której niemożliwe jest plastyczne odkształcenie materiału.
Zatem siła tarcia
To = µ
(
1 +
po
p 'o
)
N
(4.39)
Dla metali plastycznych rzeczywista powierzchnia styku Sr jest wprost proporcjonalna do obciążenia normalnego N. Wartość współczynnika tarcia µ nie zależy od obciążenia normalnego N ani od rodzaju chropowatości powierzchni tarcia. Dla metali sprężystych pole rzeczywistej powierzchni styku Sr nie jest proporcjonalne do obciążenia normalnego N, lecz zależy od rodzaju chropowatości.
Z powyższej analizy wynika, że podana hipoteza również nie uwzględnia własności fizycznych materiału trących się powierzchni. Duże wartości współczynnika tarcia stykanych płytek wzorcowych (duża gładkość powierzchni), wynikające z oddziaływania sił adhezji, są uzasadnione teoretycznie hipotezą Deriagina. Hipoteza Deriagina uwzględnia siły przyciągania między atomami, ale nie uwzględnia niszczenia powierzchni tarcia (ścieranie, sczepianie, odkształcenia, nierówności). Opisany mechanizm tarcia jest słuszny przy idealnym ślizganiu.
4.1.5.3. Adhezyjno-mechaniczna teoria tarcia suchego
W celu uwzględnienia w teorii tarcia zgodności zasad teoretycznych z wynikami badań sformułowano mechaniczno-adhezyjną teorię tarcia suchego. Autorem tej teorii jest Kragielski. W teorii tej Kragielski siły adhezji utożsamia z siłami oddziaływania molekularnego. Założył on, że siły tarcia występują w dwu postaciach. A mianowicie na rzeczywistych trących powierzchniach styku występuje mechaniczny opór ruchu, spowodowany zaczepieniem o siebie nierówności powierzchni, oraz opór spowodowany oddziaływaniem sił molekularnych, uwarunkowany wzajemnym przyciąganiem atomów lub cząsteczek trących ciał.
Dla ułatwienia rozważań wprowadzono pojęcie jednostkowej siły tarcia Fj działającej na jednostkowe pole rzeczywistej powierzchni styku. Całkowita, mierzona eksperymentalnie, siła tarcia jest równa iloczynowi powierzchni Sr przez sumę jednostkowych sił tarcia
n
To = Sr
∑
Fji
(4.40)
1
Jednostkową siłę tarcia współoddziaływania molekularnego oblicza się z zależności podanej przez Deriagina
Fj mol = a1 + b1 q
(4.41)
gdzie: a1, b1 - stałe współczynniki charakteryzujące opór materiału na ścinanie, q - rzeczywisty nacisk jednostkowy.
Jednostkową siłę tarcia wywołaną oporami mechanicznymi można przedstawić wg Bridgmana w postaci
Fj mech = a2 + b2 q
(4.42)
gdzie: a2 - składowa jednostkowej siły tarcia pochodząca od sił molekularnego wzajemnego oddziaływania na jednostkę powierzchni, b2 - współczynnik zależny od molekularnej chropowatości.
W obu przypadkach jednostkowe siły tarcia są wyrażone za pomocą identycznych zależności. Dlatego jednostkową siłę tarcia przy molekularno-mechanicznym oddziaływaniu trących powierzchni można wyrazić zależnością
Fj = Fj mol + Fj mech = a + b q
(4.43)
Na poszczególnych trących powierzchniach styku występuje mechaniczne lub molekularne oddziaływanie kojarzonych elementów maszyn.
Składowa siły tarcia wywołana oddziaływaniem molekularnym jest wyrażona zależnością
Tmol = Fj mol Sr 1
(4.44)
a składowa siły tarcia wywołana oddziaływaniem mechanicznym
Tmech = Fj mechl Sr 2
(4.45)
gdzie: Sr 1 - powierzchnia styku, na której występują współoddziaływania molekularne, Sr 2 - powierzchnia styku, na której występują współoddziaływania mechaniczne.
Rzeczywista powierzchnia styku, na której występuje współoddziaływanie molekularne i mechaniczne
Sr = Sr 1 + Sr 2
(4.46)
Całkowita siła tarcia
T = a Sr + b N
(4.46)
Współczynnik tarcia µ można określić z zależności
µ =
T
N
=
a Sr
N
+ b
(4.48)
Kragielski przyjmuje, że Sr 1 = n Sr 2, gdzie n = const. Wtedy współczynniki a i b, zależne od adhezyjnych i mechanicznych własności trących ciał stałych, są określone zależnościami
a =
n a2 + a1
n +1
; b =
n b2 + b1
n +1
(4.49)
gdzie: a1, a2 oraz b1, b2 są objaśnione ze wzorami (4.41) i (4.42).
Rzeczywistą powierzchnię styku trących powierzchni, przy pominięciu ich chropowatości, można wyznaczyć z zależności Hertza. Jeśli przy obliczaniu rzeczywistej powierzchni styku uwzględnia się chropowatość trących powierzchni, wtedy należy posłużyć się modelem. Stosując na przykład model pręcików o rozkładzie liniowym, otrzymano zależność na rzeczywistą powierzchnię styku
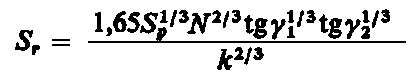
(4.50)
gdzie g1, g2 - kąty nachylenia nierówności obszaru styku.
Podana zależność jest słuszna jedynie dla liniowego rozkładu wysokości nierówności i liniowej zależności między naprężeniem a odkształceniem pręcika. Współczynnik k jest również funkcją obciążenia. W przypadku idealnie plastycznego styku (N / S ) = spl, gdzie spl granica plastyczności metalu.
Wówczas współczynnik tarcia może być wyrażony za pomocą zależności
µ =
a
spl
+ b
(4.51)
W praktyce korzystanie z podanych przez Kragielskiego zależności jest utrudnione, ponieważ w niektórych przypadkach należy uwzględniać własności odkształceń sprężystych trących metali i inne.
Zależność na współczynnik tarcia podaje Kragielski w następującej postaci
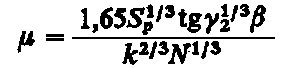
(4.52)
Z podanego wzoru wynika, że przy niezmiennych wartościach a, b, g współczynnik tarcia maleje ze wzrostem obciążenia, natomiast rośnie ze wzrostem gładkości powierzchni i ze wzrostem nominalnej powierzchni styku, a maleje ze wzrostem sprężystości materiału.
Dla dużych obciążeń pierwszy człon maleje. Wpływ czynników wchodzących do tego członu równania jest nieznaczny, a wtedy wartość współczynnika tarcia µ jest określona przez wartość współczynnika b. Dla plastycznego kontaktu trących ciał współczynnik tarcia nie zależy od obciążenia.
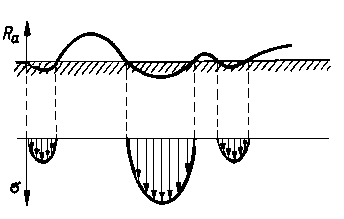
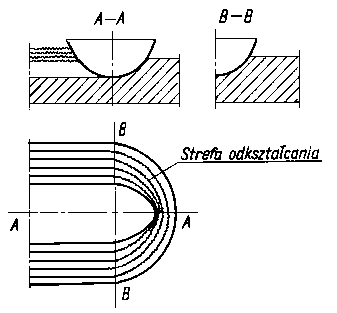
Schemat rozkładu naprężeń przy styku dwóch ciał chropowatych podano na rys. 4.13. Mechanizm tarcia można objaśnić analizując falowy charakter odkształcania materiału przez zagłębianie się nierówności powierzchni twardszego materiału w miększym (rys. 4.14).
Kragielski oparł taką interpretację na badaniach ślizgającej się korundowej igły po plastycznym tworzywie sztucznym. Materiały te nie ulegają sczepieniu, a mimo to po każdym przejściu igły po tej samej drodze (wyciśnięta bruzda nie zmieniała w zasadzie swojego kształtu) wartość współczynnika tarcia ulegała zmianom.
Rys. 4.13. Model rozkładu naprężeń w obszarze styku dwóch ciał chropowatych
Rys. 4.14. Model falowego odkształcenia metalu w obszarze zagłębionego występu nierówności
Proces takiego odkształcenia materiału polega na przemieszczaniu fali materiału przed występem oraz odsuwanie materiału na boki z bruzdy zakreślonej występem nierówności.
Jeśli odkształcenie ma charakter plastyczny, to spiętrzony przed występem materiał jest rozpłaszczony w czasie przejścia występu przez spiętrzenie, a materiał usunięty na boki z bruzdy tarcia jest ponownie przemieszczany przy przejściu następnego występu nierówności.
Opór takiego falowego odkształcenia materiału zależy od własności mechanicznych i adhezyjnych badanego materiału. Wysokość fali odkształconego materiału przed rysującym go występem i wielkość odkształconej strefy są tym większe im większa jest siła adhezji, a mniejsza granica plastyczności materiału. Falowe odkształcenie materiału podczas tarcia może mieć również sprężysty charakter, przy którym straty energii i opór tarcia są związane z histerezą materiału.
W obu przypadkach mechanizm tarcia jest określony zjawiskami mechanicznymi i adhezją metali trących. Omawiane dotychczas teorie tarcia suchego dotyczyły zjawisk tarcia w obszarach styku ślizgających się ciał.
4.1.6. Zjawiska w strefie kontaktu ciało stałe-ciało stałe
4.1.6.1. Adhezja
Adhezja jest zjawiskiem powierzchniowym, polegającym na sczepieniu się powierzchni stykających się ciał wskutek oddziaływania między nimi pola sił. Pole sił wytwarzane przez ładunki atomów (jonów, cząsteczek), z których jest zbudowana warstwa wierzchnia ciał, maleje wykładniczo wraz ze wzrostem odległości od powierzchni. Praktycznie oddziaływanie powierzchniowe typu Van der Waalsa zanika powyżej odległości 1-2 nm, dlatego dla zaistnienia sczepienia adhezyjnego jest konieczne odpowiednie zbliżenie powierzchni. Dla uzyskania dużej rzeczywistej powierzchni styku stosuje się polerowanie.
Oprócz oddziaływań typu elektrostatycznego, Lifszyc rozważa ponadto oddziaływanie typu elektrodynamicznego.
Ze względu na to, że wszystkie ciała stałe i ciekłe mają powierzchniowe pole sił, dlatego powinny one podlegać zjawisku adhezji. Wiadomo jednak, że niektóre ciała w zetknięciu ze sobą nie wykazują skłonności adhezyjnych. Dotychczas nie ma racjonalnego wytłumaczenia tego zjawiska. Należy przypuszczać, że dużą rolę w zjawiskach adhezyjnych odgrywa zgodność budowy geometrycznej i rozmieszczenie atomów (jonów) powierzchniowych nośników ładunków elektrostatycznych. W przypadku niezgodności budowy zbliżonych do siebie powierzchni adhezja nie następuje. Dużą rolę przypisuje się, zwłaszcza przy adhezji polimerów, obecności grup polarnych. Ciała o budowie polarnej są bardziej podatne do sczepiania adhezyjnego.
Dotychczas nie ma jednoznacznie ustalonych i ogólnie uznanych poglądów na istotę adhezji. Istnieje na ten temat kilka teorii. Podane wyjaśnienie adhezji oddziaływaniem sił międzycząsteczkowych jest treścią tzw. absorpcyjnej teorii adhezji. Deriagin wraz ze współpracownikami opracował tzw. elektryczną teorię adhezji.
Opierając się na wynikach uzyskiwanych przy rozrywaniu sczepionych adhezyjnie ciał, stwierdził on, że siły międzycząsteczkowe działają do odległości około 2 nm, podczas gdy przyciąganie było obserwowane przez niego na odległości do l µm. a więc 1000 nm. Uważa on, że oprócz oddziaływań międzycząsteczkowych zasadniczą rolę przy sczepieniu odgrywa podwójna warstwa elektryczna, jaką wytwarza się przy zetknięciu ze sobą dwóch ciał. Przy adhezyjnym sczepieniu polimerów, mających grupy polarne, wytworzenie elektrycznej warstwy podwójnej przypisuje się mechanizmowi donorowo-akceptorowemu.
Przy rozrywaniu sczepień adhezyjnych następuje emisja elektronów. Morozowa w swoich pracach stwierdziła prostą zależność między prędkością emitowanych elektronów a wartością adhezji. Przy rozrywaniu sczepień adhezyjnych następuje zanikanie ładunków (rekombinacja) wskutek efektu tunelowego i emisji elektronów, odprowadzania ładunków w wyniku przewodności elektrycznej powierzchni oraz wskutek wyładowań elektrycznych w warstwie gazu przylegającego do powierzchni.
Jak widać z powyższych rozważań teoria adsorpcyjna i elektryczna nie wykluczają się nawzajem, a teoria elektryczna wskazuje jedynie na źródło dodatkowych sił adhezyjnych.
W badaniach sczepiania adhezyjnego polimerów oraz procesów klejenia uznano istniejące teorie za niewystarczające i zaproponowano dyfuzyjną teorię adhezji. Autorem tej teorii jest Wojucki. Według tej teorii cząsteczki (lub ich fragmenty) zetkniętych ze sobą polimerów dyfunduje w warstwy przypowierzchniowe, przez co rozmywa się ostra granica rozdziału pomiędzy polimerami, a tworzy się przejściowo warstewka o mieszanym składzie, silnie spajająca obydwa polimery.
Istnienia takiej warstwy przejściowej eksperymentalnie nie stwierdzono, ale wprowadzenie takiego wyjaśnienia dobrze tłumaczy wpływ niektórych czynników technologicznych procesu klejenia (wpływ rozpuszczalników kleju, wpływ plastyfikatorów itd.). Trudno sobie natomiast wyobrazić dyfuzje cząsteczek przy sczepieniu adhezyjnym polimeru ze szkłem czy metalem. Natomiast eksperymentalnie stwierdzono dyfuzję metali przy występowaniu adhezyjnych zrostów międzymetalicznych zarówno w warunkach statycznych jak i dynamicznych.
Przy połączeniach adhezyjnych typu polimer-polimer lub polimer-metal uważa się, że czysta warstwa leżąca na granicy faz jest najsłabszym obszarem spojonych dwóch ciał (weak boundary layer), gdyż dyfundują do niej związki o najmniejszej masie cząsteczkowej.
W niektórych typach polimerycznych połączeń adhezyjnych może następować tworzenie kompleksów z przeniesieniem ładunku, co zwiększa trwałość utworzonego połączenia. Zmiany skłonności ciał do adhezji można uzyskiwać poprzez wstępną obróbkę chemiczną ciał mających tworzyć połączenie adhezyjne (np. pokrywanie warstewką substancji o dużej skłonności adhezyjnej). Zjawiska adhezyjne mają duże znaczenie w badaniach tarciowych, gdyż są one źródłem znacznej części oporów tarcia, zwłaszcza przy tarciu suchym.
W Związku Radzieckim dla określenia składowej adhezyjnej (molekularnej) oporów tarcia została przyjęta znormalizowana metodyka, w której siłę sczepienia adhezyjnego określa się dla polerowanych płytek wykonanych z badanych metali.
Należy zaznaczyć, że przy kontakcie ciał stałych adhezja może wystąpić jedynie w mikroobszarach rzeczywistego styku. Ogromny wzrost sczepności uzyskuje się przez wprowadzenie między kontaktujące się powierzchnie cieczy (np. oleju smarnego), gdyż powoduje to wzrost rzeczywistej powierzchni styku, o ile oczywiście ciecz zwilża obie powierzchnie stałe. Przykładem może tu być sczepienie dwóch płytek szklanych, zwilżonych cieczą.
Zjawisko adhezji metali, jak i cieczy smarnej do metali, może być jedną z przyczyn tarcia skokowego (szarpanego), tzw. „stic-slip" (zatrzymanie - poślizg). Tarcie skokowe występuje zazwyczaj wówczas, gdy w skojarzeniu trącym styka się powierzchnia chwilowo nieruchoma z powierzchnią ciała poruszającego się względem tej powierzchni. Warunkiem koniecznym do wystąpienia stic-slip jest wyższa wartość współczynnika tarcia spoczynkowego od współczynnika tarcia kinetycznego oraz stosunkowo mała prędkość względna i duże obciążenia.
Zjawisko stic-slip polega na przerywanym ruchu elementów skojarzenia tarciowego oraz powstawaniu w nim drgań. Drgania te często przenoszą się na podzespoły, a nawet na całe maszyny czy pojazdy. Badania tarcia typu stick-slip podjęto głównie ze względu na występowanie tego zjawiska w przekładniach hydrokinetycznych pojazdów i maszyn roboczych.
4.1.6.2. Zjawiska adhezyjno-dekohezyjne
Przy wzajemnym kontakcie dwóch ciał stałych występuje styk powierzchniowy. Rozróżnia się kontakt ciał nieruchomych i kontakt ciał przemieszczających się względem siebie. Przy kontakcie idealnie gładkich powierzchni, rzeczywista powierzchnia styku byłaby równa nominalnej powierzchni styku. Ponieważ realna powierzchnia styku ma mnóstwo nierówności i styk między powierzchniami występuje jedynie na nierównościach, dlatego też rzeczywista powierzchnia styku stanowi jedynie niewielki ułamek powierzchni nominalnej. Jak już wspomniano na powierzchni ciała występuje pole sił, którego oddziaływanie na znajdujące się w jego zasięgu ciała powoduje przeciąganie - adhezję. Powiązanie adhezyjne jest tym silniejsze, im silniejsze jest oddziaływanie elektrostatyczne i elektrodynamiczne pomiędzy zetkniętymi ze sobą ciałami. Wiąże się to ściśle z budową ciał, a więc i ich powierzchni. Siły adhezyjne mają charakter addytywny, dlatego też im większa jest rzeczywista powierzchnia styku, tym silniejsze występuje sczepienie adhezyjne. Oddziaływanie adhezyjne maleje bardzo gwałtownie (wykładniczo) wraz ze wzrostem odległości między kontaktującymi się ciałami.
Należy zaznaczyć, że siły adhezyjne, a więc siły działające na powierzchni ciała są to takie same siły, jakie działają we wnętrzu ciała. We wnętrzu ciała spełniają one funkcję spajającą elementy budowy ciała w jedną całość, a więc są to siły spójności czyli siły kohezji, natomiast na powierzchni ciała ze względu na brak wysycenia sił od zewnątrz spełniają one rolę sił adhezyjnych.
Wszelkie oddziaływania prowadzące do odrywania elementów budowy (lub ich zespołów) od ciała, a więc działania dekohezyjne napotykają opór sił spójności (kohezyjnych), natomiast odrywanie lub przemieszczanie względem siebie ciał zetkniętych ze sobą napotyka opór sił adhezyjnych.
Gdybyśmy przemieszczali względem siebie ciała kontaktujące się powierzchniami idealnie gładkimi, wówczas napotkalibyśmy jedynie opór sił adhezyjnych.
Przemieszczanie względem siebie rzeczywistych powierzchni styku, a więc powierzchni mających mnóstwo nierówności, napotyka nie tylko opór adhezyjny, lecz dodatkowym źródłem oporu jest przemieszczanie się względem siebie nierówności obu kontaktujących się ze sobą powierzchni. Powoduje ono odkształcenia sprężyste i plastyczne, bruzdowanie, ścinanie nierówności, tworzenie rys itp. Wszystkie te oddziaływania występują wbrew siłom spójności, a więc są to działania dekohezyjne.
Na opory tarcia przy przemieszczaniu się względem siebie dwóch ciał stałych składają się więc opory adhezyjne (Tadh i opory kohezyjne Tkoh)
Tt = Tadh + Tkoh
Jest to najogólniejsze prawo trybologiczne. W zależności od rodzaju i warunków tarcia poszczególne człony mogą wzrastać lub maleć, albo nawet zupełnie zanikać. Tak np. przy tarciu wewnętrznym cieczy człon adhezyjny jest zbliżony do zera, natomiast przy tarciu zewnętrznym idealnie gładkich powierzchni człon kohezyjny byłby równy zeru. Oprócz wymienionych tu skrajnych przypadków, w których jeden z członów zanika, istnieje wiele przypadków pośrednich, w których obydwa człony -adhezyjny i kohezyjny-mają znaczną wartość. Powyższe ujęcie ma duże znaczenie ze względów pojęciowych, natomiast dokładne obliczenie wartości poszczególnych członów jest trudne. Istniejące wzory pozwalają na obliczanie zasadniczych elementów obu członów, a stopniowe poznawanie zjawisk trybologicznych umożliwi w przyszłości zwiększanie dokładności obliczeń.
Opierając się na powyższym ujęciu możliwe jest prognozowanie i wyjaśnianie niektórych zjawisk trybologicznych. Tak np. wiadomo, że wartość współczynnika tarcia wraz ze wzrostem gładkości powierzchni zmniejsza się, ale przy bardzo dużym stopniu gładkości jego wartość zaczyna gwałtownie wzrastać. Przy wygładzaniu zmniejszamy wartość członu kohezyjnego, gdyż zmniejsza się liczba nierówności, które podlegają ścinaniu czy odkształceniom, natomiast stopniowo zaczyna wzrastać rzeczywista powierzchnia styku i rośnie wartość członu adhezyjnego, a wraz z nim wartość współczynnika tarcia.
W powyższym świetle można również rozważyć przydatność poszczególnych teorii tarcia. Po bliższym zapoznaniu się z nimi można stwierdzić, że wszystkie one są słuszne, ale jednak w pewnym zakresie, tj. każda z nich rozpatruje proces tarcia zbyt jednostronnie i wskutek tego nie może być w pełni słuszna. Mechaniczne teorie tarcia uwzględniają przede wszystkim człon kohezyjny, tj. w mniejszym lub większym stopniu uwzględniając ujęcie matematyczne zjawisk odbywających się wbrew siłom kohezyjnym (odkształcenie sprężyste, odkształcenie plastyczne, ścinanie itd.). Molekularne teorie tarcia główną uwagę poświęcają oddziaływaniu adhezyjnemu i właśnie na tym polega ich jednostronność. Należy zaznaczyć, że nazwa teoria molekularna w aspekcie oddziaływań adhezyjnych nie jest słuszna, gdyż adhezję dwóch powierzchni możemy rozpatrywać jako zjawisko makro, będące jednak sumą oddziaływań przyciągania w sferze molekularnej. Należy zdać sobie sprawę z tego, że to co poszczególni autorzy uważają za mechaniczne przyczyny tarcia jest też sumą molekularnych oddziaływań kohezyjnych.
Tak więc określenie molekularne może się odnosić zarówno do członu kohezyjnego, jak i adhezyjnego. Niektórzy autorzy dużo uwagi poświęcają teoriom energetycznym, przypisując im uprzywilejowaną rolę. Tymczasem jest zupełnie obojętne czy opory tarcia są rozpatrywane jako reakcja na siły ścinające, czy też całe to zjawisko rozważamy w kategoriach energetycznych, albo też określimy ilość pracy, jaką trzeba doprowadzić do układu, aby te opory pokonać. W każdej z teorii chodzi o te same oddziaływania, a zmienia się tylko podejście do rozwiązania zagadnienia. Zachodzi pytanie jak w powyższy sposób rozumowania włączyć oddziaływanie całego zespołu zjawisk ubocznych, towarzyszących tarciu, a mających duży wpływ na wartość oporów przemieszczania powierzchni względem siebie. Weźmy, jako przykład, zjawiska elektryczne towarzyszące tarciu. Powodują one zwiększenie lub zmniejszenie potencjału kontaktujących się ze sobą powierzchni, a więc wpływają na zmiany intensywności przyciągania się tych dwóch powierzchni, czyli powinny być uwzględnione jako element składowy członu adhezyjnego. Drugim przykładem niech będą drgania powstające jako efekt uboczny procesu tarcia. Powodują one cykliczne odchylenia od położenia równowagi, a więc oddziałują przeciw siłom kohezyjnym. Osobnym, bardzo specyficznym przypadkiem jest ścinanie połączeń adhezyjnych przy zetknięciu się dwóch mikronierówności. W wyniku oddziaływania adhezyjnego może ono w pewnych warunkach (bardzo duże zbliżenie, wysoka temperatura itp.) doprowadzić do powstania (w tym miejscu styku) jednego ciała stałego. W miejscu tym siły adhezyjne zamieniają się na siły kohezyjne i chcąc obliczyć opory ścinania połączeń adhezyjnych, musimy stosować wzory właściwe do obliczania elementów członu kohezyjnego. Paradoksalne jest to jedynie pod względem terminologicznym, natomiast jest słuszne pod względem logicznym. Przystępując do rozpatrzenia zjawisk występujących przy kontaktowaniu się ze sobą dwóch ciał stałych należy stwierdzić, że jeżeli będzie to kontakt metal-metal, to opory tarcia są zawsze duże. Duże siły spójności przy więzi metalicznej sprawiają, że metale w porównaniu z innymi rodzajami ciał stałych (z wyjątkiem ciał o wiązaniu kowalentnym) mają dużą wytrzymałość na odkształcenia sprężyste i plastyczne, dlatego też przy występowaniu nierówności powierzchniowych człon kohezyjny ma bardzo dużą wartość. Wygładzanie nierówności na powierzchni metalu doprowadza do zmniejszania się wartości członu kohezyjnego, a do wzrostu wartości członu adhezyjnego. Ze względu na istnienie ładunków powierzchniowych (dodatnio naładowane zręby atomowe i ujemnie naładowany „gaz" elektronowy) człon adhezyjny może przybierać bardzo wysokie wartości. Należy zaznaczyć, że dla technicznie czystych powierzchni metali zasadniczą rolę odgrywa człon kohezyjny, natomiast człon adhezyjny, ze względu na dużą chropowatość oraz procesy sorpcyjne (tworzenie tlenków, adsorpcja pary wodnej) obniżające energie powierzchni, odgrywa mniejszą rolę.
Jeżeli kontaktującymi się ze sobą ciałami w procesie trybologicznym są tworzywa sztuczne, wówczas przebiegające zjawiska mają nieco inny charakter. Polimeryczne tworzywa sztuczne są to ciała zbudowane z cząsteczek powiązanych pomiędzy sobą siłami międzycząsteczkowymi (Van der Waalsa). Dużą spójność, a więc i korzystne cechy jako tworzywa konstrukcyjne, uzyskują one w wyniku sumowania się (addytywności) niektórych rodzajów sił międzycząsteczkowych (Van der Waalsa). W przypadku cząsteczek o bardzo dużej masie cząsteczkowej, a więc cząsteczek olbrzymich, duża kohezja jest wynikiem głównie addytywności sił dyspersyjnych (Londona) oraz sił konformacyjnych. W przypadku polimerów, w skład których wchodzą pierwiastki nadające cząsteczkom polarność (np. tlen, chlor, fluor, itd.), dużą rolę mogą odgrywać ponadto siły indukcyjne (Debye'a) i orientacyjne (Kessoma). Polimery usieciowane przestrzennie, mające poszczególne podstawowe ,,cegiełki” powiązane przestrzennie wiązaniami kowalentnymi, odznaczają się szczególnie dużą spójnością. Dlatego też opory tarcia przy oddziaływaniu przeciw siłom kohezyjnym mają dużą wartość, natomiast ze względu na brak ładunków elektrycznych na powierzchni, co wynika z budowy ciała (ładunki mogą się tworzyć podczas tarcia), człon adhezyjny ma małą wartość.
Osobnym zagadnieniem jest tarcie elastomerów, a przede wszystkim gumy. Kontakt powierzchniowy powoduje objętościowe odkształcenie sprężyste. Po zakończeniu działania polegającego na wymuszaniu odkształcenia elastomer wraca do stanu nieodkształconego. Stwierdzono, że w gumie energia odkształcenia jest rozpraszana głównie przez histerezę. Tworzywa polimeryczne mają słabą zdolność do odprowadzania ciepła, toteż istnieje tendencja do kumulacji ciepła w obszarach tarcia. Może to spowodować w tworzywie zmiany odwracalne, jak jego mięknienie, topnienie (zwłaszcza w przypadku polimerów termoplastycznych) oraz zmiany nieodwracalne (degradacja mechaniczna polimeru, utlenianie lub zwęglanie). Zjawiska te mogą w zasadniczy sposób zmieniać warunki tarcia kontaktujących się powierzchni polimerów. Powierzchniowe mięknienie polimeru zwiększa gwałtownie rzeczywistą powierzchnię styku, a więc zwiększa się wartość członu adhezyjnego, natomiast zmniejsza się wartość członu kohezyjnego ze względu na zmniejszenie odporności na deformację i ścinanie. Następuje również przenoszenie materiału na kontaktującą się powierzchnię materiału o wyższej temperaturze mięknienia.
Podczas tarcia polimerów termoplastycznych powstaje również znaczna deformacja objętościowa. Polimery jako dielektryki mają skłonność do silnego elektryzowania się. Udało się ustalić szereg trybologiczny polimerów (rys. 4.15) Skłonność do elektryzowania się przy wzajemnym kontaktowaniu jest zależna od położenia polimeru w szeregu. Największy efekt elektrostatyczny uzyskuje się przy doborze pary materiałów leżących na przeciwległych końcach szeregu. Jak już wspomniano uprzednio, pojawianie się ładunków elektrostatycznych powoduje zwiększenie wartości członu adhezyjnego i zwiększenie wskutek tego ogólnych oporów. Podobne efekty uzyskuje się wraz ze zwiększeniem polarności przeciwpróbki ciernej.
Przy kontakcie ślizgowym metal-polimer człon adhezyjny ma zazwyczaj nieznaczną wartość ze względu na nieaktywność powierzchniową polimeru. Aktywność ta wzrasta wraz ze wzrostem polarności polimeru, jak np. z intensywnością
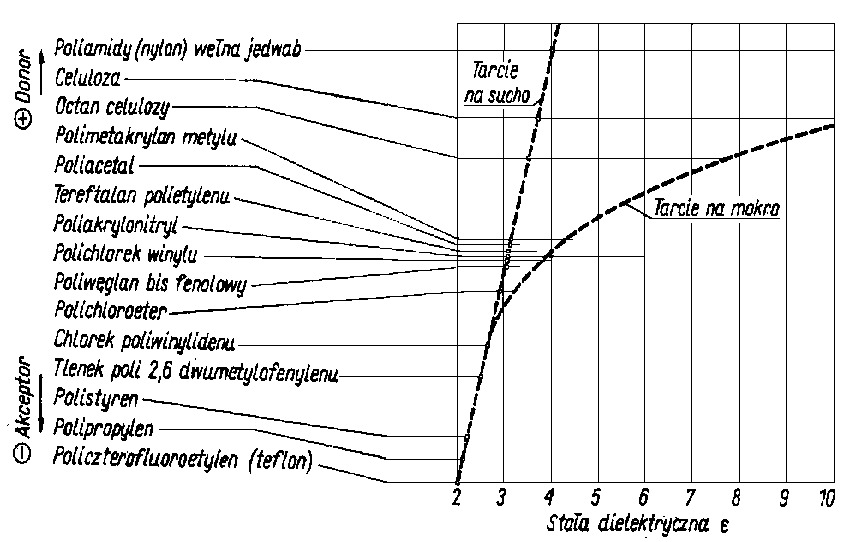
Rys. 4.15. Tryboelektryczny szereg polimerów
tworzenia się ładunków elektrostatycznych. Zaznacza się jednak znaczny wpływ metalu, a zwłaszcza jego zdolność do odprowadzania ładunku. Należy jednak podkreślić, że możliwe jest znaczne oddziaływanie adhezyjne metal-polimer przy dużej gładkości powierzchni polimeru. Tak np. w laboratorium podczas badań przeprowadzonych przez autorów stwierdzono zużywanie się stali przy jej kontakcie z przeciwpróbką z metakrylanu metylu (plexiglas) o lustrzanie gładkiej powierzchni. Powierzchnia polimeru zostaje jednak zazwyczaj stosunkowo łatwo niszczona mechanicznie (rysowanie, bruzdowanie, deformacja) i w tym przypadku człon kohezyjny ma dużą wartość. Polimery wewnętrznie spójne, mające odporną warstwę powierzchniową, w przypadku jej znacznej gładkości powodują małe sumaryczne opory tarcia, gdyż zarówno człon adhezyjny, jak i kohezyjny mają małą wartość.
4.1.7. Tarcie monokryształów
Badanie tarcia monokryształów ma na celu przede wszystkim ustalenia podstawowych praw dotyczących tarcia ciał stałych. Umożliwia ono określenia uprzywilejowanych płaszczyzn i kierunków poślizgu, powiązanie oporów tarcia z gęstością upakowania atomowego i ułożenia płaszczyzn atomowych. Odpowiednio zaprogramowane badania umożliwiają również śledzenie wpływu defektów budowy na opory tarcia monokryształów oraz powiązania ich z energią powierzchni, współczynnikiem sprężystości i twardością. Przeprowadzone badania monokryształów czystych metali i ich stopów oraz ciał krystalicznych o budowie jonowej i kowalentnej pozwalają na dokonanie pewnych uogólnień. Dla tego celu szczególnie duże znaczenie mają prace H. D. Buckley'a (12), (13), (14), (15), na których w głównej mierze oparto przedstawione rozważania. Jak wiadomo płaszczyzny poślizgu w kryształach są zazwyczaj najgęściej upakowanymi płaszczyznami i pomiędzy tymi płaszczyznami występują w krysztale największe odstępy. Jeżeli w krysztale występuje poślizg, to zazwyczaj wzdłuż kierunku o najgęstszym upakowaniu, gdyż znacznie większe naprężenie ścinające jest potrzebne dla zainicjowania poślizgu płaszczyzn o mniejszej gęstości upakowania. Pewne zaburzenia od tej reguły występują w przypadku kryształów o więzi jonowej (np. soli kuchennej).
Dla sieci ściennie centrowanej układu regularnego liczba możliwych wariantów poślizgu wynosi 12, dla sieci przestrzennie centrowanej 48, a dla sieci heksagonalnej zwartej, na płaszczyźnie podstawy, istnieją tylko trzy warianty poślizgu (rys. 4.16). Im większa liczba możliwych wariantów poślizgu, tym łatwiej
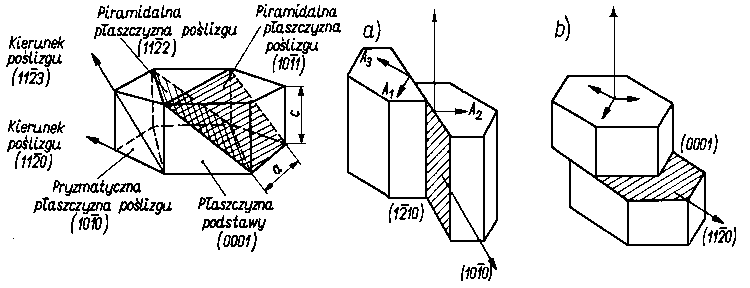
Rys. 4.16. Płaszczyzny poślizgu i warianty poślizgu dla kryształu o sieci heksagonalnej:
- - poślizg na płaszczyźnie pryzmatycznej,
- - poślizg na płaszczyźnie podstawy
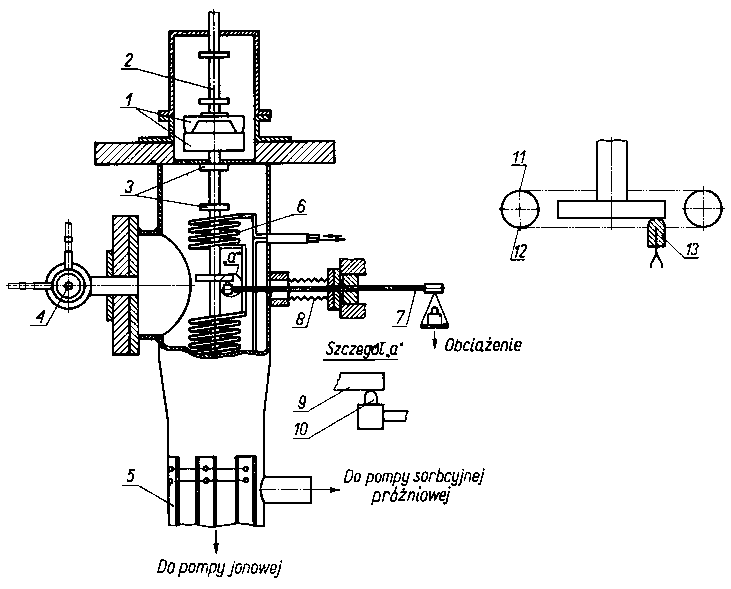
Rys. 4.17. Urządzenie do badania tarcia w wysokiej próżni;
- - napęd magnetyczny,
- - trzpień napędowy,
- - łożyska,
- - wskaźnik jonizacyjny z zimną katodą,
- - grzejniki osuszające,
- - wężownice kriogeniczne,
- - pomiar siły tarcia,
- - mieszek,
- - dysk,
- - ślizgacz,
- - spirale grzejne z drutu tantalowego,
- - uchwyt z miki dla spirali grzejnej,
- - termopara chromel-alumel osadzona w ślizgaczu
można wywołać odkształcenie plastyczne. Należy jednak zaznaczyć, że w takich warunkach zwiększa się również tendencja do występowania umocnień. Do badań tarcia monokryształów Buckley stosował urządzenie próżniowe przedstawione na rys. 4.17. Skojarzenie trące składało się z tarczy i ślizgacza wykonanego z monokryształu. Tarcza jest zamocowana na trzpieniu napędzanym dwudziestobiegunowym magnesem poprzez diafragmę. Magnes napędzający znajdował się na zewnątrz układu próżniowego. Ślizgacz umocowany był na ruchomym ramieniu przenoszącym zarówno obciążenie, jak i siłę tarcia. Dla usunięcia z powierzchni trących zaadsorbowanych substancji w układ próżniowy wmontowano działo elektronowe. Rezultaty badań zostaną przedstawione oddzielnie dla różnych typów struktur krystalicznych.
4.1.7.1. Kryształy o więzi metalicznej
Sieć ściennie centrowana układu regularnego. Dla metali krystalizujących w tej sieci stwierdzono anizotropowość oporów tarcia. Najmniejsze opory tarcia występują przy poślizgu na uprzywilejowanych kierunkach, tj. na płaszczyznach o największej gęstości atomowej. Jak widać z tabl. 4.7 zwiększenie gęstości atomowej powoduje
Tablica 4.7.
Zachowanie się monokryształów miedzi (sieć ściernie centrowana układu regularnego) podczas tarcia
Płaszczyzna Gęstość atomowa (liczba atomów/jednostkę powierzchni) Odstęp pomiędzy płaszczyznami nm Energia powierzchniowakJ
m²
Współczynnik tarcia
Powietrze 1000 hPa Próżnia 1,33 mPa
(100) 0,152 7 0,128 2 892 0,60 > 40
(110) 0,108 0 0,180 5 - 40 > 40
(111) 0,172 2 0,208 2499 21 21
zmniejszenie energii powierzchniowej, zmniejszenie współczynnika tarcia oraz zwiększenie współczynnika sprężystości.
Sieć przestrzennie centrowana układu regularnego. Dla metali krystalizujących w tej sieci należało oczekiwać raczej bardzo niewielkiego wpływu kierunku poślizgu na opory tarcia. Jednym z metali krystalizujących w tej sieci, a wykazujących małą anizotropowość własności, jest wolfram. Badania tarciowe przeprowadzone w trzech kierunkach poślizgu wykazały dosyć znaczne różnice w wartościach współczynnika tarcia w próżni. Dla płaszczyzny (100) i dla kierunku poślizgu (100) współczynnik tarcia wynosi 0,70, dla tej samej płaszczyzny i kierunków (120) i (110) wynosi 0,98 i 1,23. Tak więc i dla tego przypadku stwierdzono, że najmniejsze opory tarcia i największa twardość występują w kierunkach o największym upakowaniu atomowym.
Sieć heksagonalna. Metale krystalizujące w tej sieci odznaczają się małą skłonnością do deformacji plastycznej ze względu na małą liczbę możliwych wariantów poślizgu (tabl. 4.8). Metale o sieci heksagonalnej mają mniejszą zdolność
Tablica 4.8.
Płaszczyzny i kierunki poślizgu różnych substancji krystalicznych (wg Buckleya)
Typ struktury krystalicznej Płaszczyzna poślizgu Kierunek poślizgu Liczba możliwych wariantów poślizgu Przykłady
Kryształy o więzi metalicznej
Sieć ściennie centrowana układu regularnego (111) (110) 12 Cu, Ni, Al, Ag, Au
Sieć przestrzennie centrowana układu regularnego (110)
(112)
(123) (111)
(111)
(111) 12
12
24 Fe, W, Mo
Sieć heksagonalna zwarta - poślizg na płaszczyźnie: bazowej pryzmatycznej piramidalnej (0001)
(1010)
(1011)
(1120)
(1120)
(1120)
3
3
6
Co Be Re Cd Zn La Ti Zr Hf
Kryształy o więzi jonowej
Sieć regularna (sól kuchenna)
Sieć romboedryczna (110)
(0001) (110)
(1120) 12
3 MgO, LiF, NaCl, KBr, Al2O3
Kryształy o więzi kowalencyjnej
Diament (111) (110) 12 Si, Ge, diament
do adhezji, powodują mniejsze opory tarcia oraz mniejsze zużycie niż metale układu regularnego. Wyraźnie mniejsza jest również skłonność do zacierania. Uwydatnia się u nich wyraźna anizotropowość własności, między innymi również własności tarciowych. Badania tarcia monokryształów metali krystalizujących w sieci heksagonalnej, jak kobaltu, berylu i renu (tabl. 4.9) wykazały, że najmniejsze opory tarcia występują przy poślizgu w płaszczyźnie podstawy (0001). W tym kierunku występuje również największa gęstość upakowania atomowego oraz najniższy współczynnik tarcia. Ze względu na to, że poślizg w płaszczyźnie podstawy jest możliwy w trzech kierunkach, współczynnik tarcia przyjmuje tę samą wartość, co przy kącie 60° ułożenia płaszczyzny podstawy monokryształu do powierzchni tarcia.
Tablica 4.9.
Wartości współczynników tarcia dla metali krystalizujących w sieci heksagonalnej (wg Buckleya)
Płaszczyzna poślizgu (0001) (1010)
Kierunek poślizgu (1120) (1010) (1120) (0001)
Kobalt 0,38 - 0,80 -
Beryl 0,48 0,51 - 0,70
Ren 0,28 0,38 0,50 0,80
Tytan 0,56 - 0,36 -
Dla berylu przeprowadzono badania wpływu wartości kąta utworzonego pomiędzy płaszczyzną podstawy monokryształu a powierzchnią tarcia na wartość współczynnika tarcia. Otrzymane wyniki przedstawione zostały na rys. 4.18.
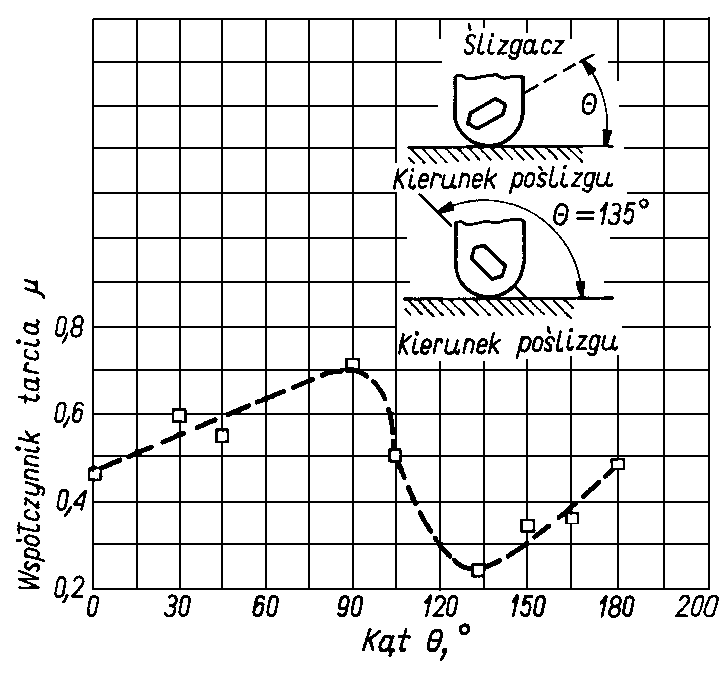
Rys. 4.18. Współczynnik tarcia dla monokryształu berylu trącego o polikrystaliczną tarczę berylową w próżni przy różnych kątach Q między płaszczyzną podstawy a płaszczyzną powierzchni (obciążenie 2,50 N prędkość poślizgu 0,13 mm/s, ciśnienie 1333 nPa bez podgrzewania zewnętrznego próbki)
Jak widać z rysunku współczynnik tarcia ma największą wartość przy prostopadłym (kąt 90°) ułożeniu płaszczyzny podstawy monokryształu berylu do powierzchni tarcia. Interesujący jest fakt, że najmniejszej wartości współczynnika tarcia nie obserwowano przy równoległym ułożeniu płaszczyzny podstawy monokryształu do powierzchni tarcia, lecz przy kącie 135°. Dla tytanu występują mniejsze opory tarcia w płaszczyźnie pryzmatycznej (1010) niż w płaszczyźnie podstawy (0001), gdyż (w odróżnieniu od innych metali tej grupy) płaszczyzna podstawy nie jest płaszczyzną najgęściej upakowaną i większą gęstość upakowania ma płaszczyzna pryzmatyczna (1010).
4.1.7.2. Kryształy o więzi jonowej
Dla kryształów o więzi jonowej zachowanie w procesach trybologicznych jest uwarunkowane siłą oddziaływania jonowego między elektrododatnim jonem metalu i silnie elektroujemnym jonem chlorowca, tlenu itp. Kryształy tego typu mają bardzo ograniczone zastosowania trybologiczne, dlatego też badaniami obejmowane są dla uzyskania poglądu na ich zachowanie się podczas procesów tarcia różnych grup substancji krystalicznych. Stosunkowo najbardziej zainteresowano się krystaliczną budową tlenku glinu - szafiru, który stosowano jako materiał na łożyska w różnego rodzaju przyrządach precyzyjnych. Stwierdzono, że szafir wykazuje anizotropię zachowania się podczas tarcia zbliżoną do metali krystalizujących w układzie heksagonalnym. W badaniach tarcia szafiru stwierdzono, że przy ruchu postępowo-zwrotnym występują różne opory i intensywność zużywania jest zależna od kierunku ruchu(rys. 4.18, tabl. 4.10). Zjawisko to niektórzy uzasadniają budową kryształu szafiru podobną do rozsuniętej nieco talii kart, co powoduje różne zachowanie się w zależności od kierunku ruchu. Kryształy jonowe krystalizujące w układzie regularnym
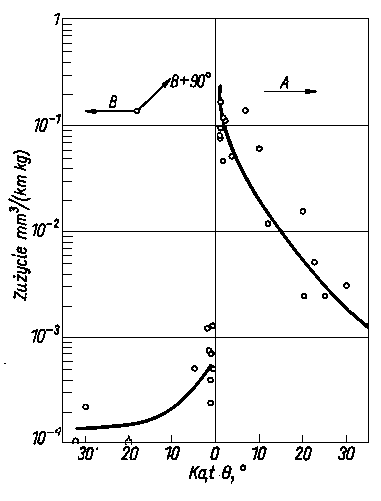
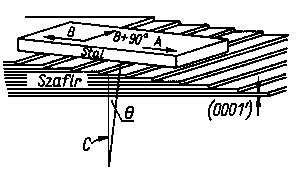
Rys. 4.19. Zależność intensywności zużywania kryształu szafiru od kąta Q i kierunku tarcia;
- - kierunek intensywnego zużywania,
- - kierunek małego zużywania,
- +90° - kierunek o pośredniej intensywności zużywania (wg Duwella)
Tablica 4.10.
Wartości współczynników tarcia monokryształów szafiru 1)
Płaszczyzna poślizgu Współczynnik tarcia Kierunek poślizgu
(0001) (1120) 0,50
(1010) 0,96
(1010) (1120) 0,93
(0001) 1,00
1) Warunki: obciążenie 10 N, v = 0,013 cm/s, temperatura 20°C, ciśnienie 13,3 nPa.
zachowują się zupełnie inaczej niż metale krystalizujące w tym samym układzie. Tak np. dla soli kuchennej płaszczyzną o największej gęstości atomowej jest płaszczyzna (100), a płaszczyzną o najłatwiejszym poślizgu - płaszczyzna (100). W przypadku Nad naprężenie ścinające jest w płaszczyźnie (100) cztery razy większe niż w płaszczyźnie (110), a dla LiF-sześć razy większe (w temperaturze 100°C). Zjawiska te wynikają z oddziaływań międzyjonowych.
4.1.7.3. Kryształy o więzi kowalentnej
Elementy budowy kryształów tego typu są powiązane pomiędzy sobą silnymi wiązaniami kowalentnymi. W przypadku diamentu występuje koordynacja tetraedryczna atomów węgla. Jak wykazały badania najmniejsze opory tarcia w przypadku diamentu występują w płaszczyźnie najgęstszego upakowania atomów (tabl. 4.11).
Tablica 4.11.
Wartości współczynników tarcia dla diamentu (wg Buckleya)
Płaszczyzna poślizgu Kierunek poślizgu Energia powierzchniowa kJ/m² Współczynnik tarcia
przy 1000 hPa przy 13,3 nPa
(100) (100) 7050 0,15 -
(110) - 0,05 -
(111) -1) 3500 0,05 0,9
1) Kierunek na płaszczyźnie (111) nie ma wpływu na współczynnik tarcia.
Porównanie zachowania się podczas tarcia substancji mono- i polikrystalicznych. Na podstawie przeprowadzonych badań tarcia monokryształów miedzi i miedzi polikrystalicznej można stwierdzić, że wskutek występowania w materiale granic ziarn następuje wyraźne zwiększenie wartości współczynnika tarcia w porównaniu z wartością tego współczynnika dla monokryształu. Jeżeli tarcie materiału polikrystalicznego prowadzi się w warunkach, w których jest możliwe steksturowanie, wówczas następuje zmniejszenie wartości współczynnika tarcia.
Stopy metali znajdujące się w warunkach, w których jest możliwe przejście ze stanu nieuporządkowanego do stanu uporządkowanego, tj. o równomiernym rozłożeniu jednego składnika stopowego w drugim, charakteryzuje się mniejszą wartością współczynnika tarcia. Wzrost nieuporządkowania zwiększa współczynnik tarcia, powoduje zmniejszenie twardości i modułu sprężystości.
Ogólnie należy więc stwierdzić, że najmniejsze opory tarcia występują w płaszczyznach najgęstszego upakowania atomów. Wyjątek stanowią tu niektóre kryształy jonowe, w których w płaszczyznach najgęstszego upakowania mogą występować silne oddziaływania międzyjonowe.
4.2. Tarcie wewnętrzne ciał stałych. Smary stałe
4.2.1. Budowa i tarcie wewnętrzne smarów stałych
Tarcie zewnętrzne ciał stałych charakteryzuje się z reguły znacznymi oporami. W celu zmniejszenia oporów tarcia wprowadza się między współpracujące powierzchnie smar, tj. substancję o małych oporach tarcia wewnętrznego, przez co zastępuje się tarcie zewnętrzne (suche) ciał stałych tarciem wewnętrznym. Do smarowania stosuje się zazwyczaj smary ciekłe (oleje) lub smary plastyczne. Smary te mają jednak ograniczoną odporność mechaniczną i cieplną. Znacznie większą odporność mechaniczną i cieplną mają smary stałe. Nazwą tą określa się ciała stałe odznaczające się stosunkowo małymi siłami spójności i dające się w związku z tym łatwo odkształcać plastycznie. Mała spójność może charakteryzować albo całe ciało, albo też może ona występować tylko w pewnych uprzywilejowanych kierunkach poślizgu, a więc wtedy ciało pod względem spójności ma charakter anizotropowy. Anizotropowością
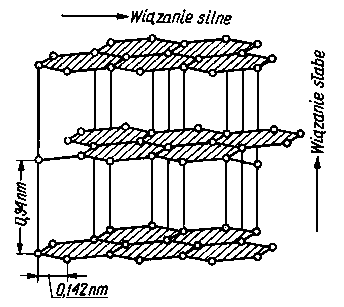
Rys. 4.20. Siatka krystaliczna grafitu
Rys. 4.21. Siatka krystaliczna dwusiarczku molibdenu MoS2:
- - widok z góry,
- - widok z boku
sił spójności odznaczają się przede wszystkim ciała stałe o warstwowej budowie siatki krystalicznej.
Warstwowa budowa siatki krystalicznej występuje wówczas, gdy w węzłach siatki znajdują się małe dodatnie jony i duże ujemne jony, łatwo polaryzowalne. W ten sposób krystalizują np. siarczki, selenki i niektóre halogenki metali. Warstwową budowę siatki krystalicznej mają również grafit i azotek boru. W związkach tych atomy ułożone w płaskiej warstwie są związane pomiędzy sobą wiązaniami kowalentnymi, a więc silnymi, natomiast pomiędzy warstwami występuje znacznie słabsza więź typu elektrostatycznego. Związki krystaliczne o budowie warstwowej odznaczają się wyraźnie zaznaczonymi płaszczyznami poślizgu i płaszczyznami łupliwości, które biegną wzdłuż poszczególnych warstw.
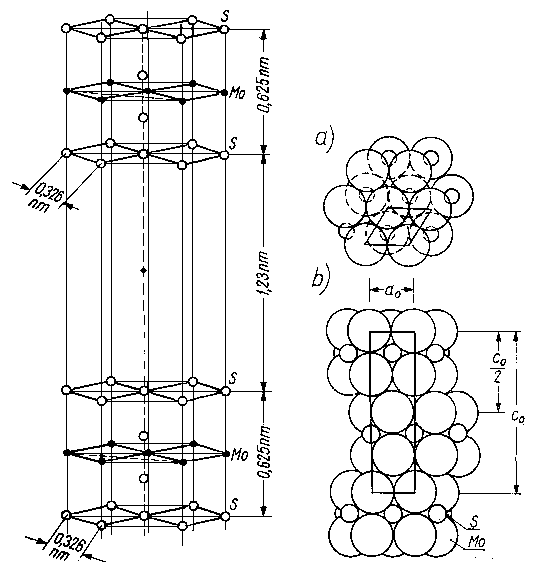
Na rysunkach 4.20 do 4.24 przedstawiono przykładowo strukturę krystaliczną kilku substancji, w przypadku których wyraźnie zaznacza się anizotropowość własności spójnościowo-mechanicznych.
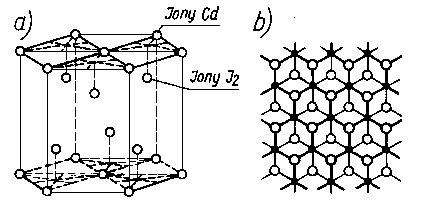
Rys. 4.22. Siatka krystaliczna jodku kadmu CdJ2:
- widok z boku,
- widok z góry
Rys. 4.23. Siatka krystaliczna wodorotlenku kadmu Cd(OH)2:
- widok z góry,
- widok z boku,
- struktura heksagonalna gęsto upakowana, utworzona przez atomy tlenu,
- prosta struktura heksagonalna utworzona przez atomy metalu (kadmu)
(a)
Rys. 4.24. Siatka krystaliczna selenku niobu Nb2Se5:
- widok z góry,
- widok z boku
(b)
Jak widać z rysunków atomy metalu ułożone w siatce krystalicznej są oddzielone podwójną warstwą atomów tlenowców (tlen, siarka, selen) lub chlorowców (chlor, brom, jod). Atomy tlenowców są ułożone bezpośrednio jeden nad drugim. Stwarza to określone sytuacje koordynacji jonowej oraz płaszczyzn symetrii. Małe opory tarcia wewnętrznego wykazują tylko kryształy o określonej budowie. Ten sam związek, mogący krystalizować w kilku odmianach polimorficznych, może tworzyć typ kryształów zupełnie nieprzydatny jako smar stały. Podobnie zachowują się roztwory stałe o różnym wzajemnym stosunku substancji krystalizujących samodzielnie w postaci kryształów warstwowych. Nieodpowiedni wzajemny stosunek składników roztworu stałego może powodować zaburzenia w strukturze utworzonej siatki krystalicznej stopu, co wiąże się ze zmianą charakteru spójności otrzymanego kryształu stopu.
Zagadnienia te zostały obszernie przeanalizowane w pracy Jamisona i Cosgrovego (26). Stwierdzili oni, że na spójność kryształów zasadniczy wpływ ma wzajemny stosunek wymiarów krystalograficznych a„ i c, (patrz rys. 4.25 do 4.27), zwany przez wspomnianych autorów stosunkiem osiowym. Odpowiednie wartości stosunku osiowego dają albo dobre, albo złe zachowanie się substancji jako smaru stałego. Zmiany własności smarnych rozważają oni opierając się na modelu sztywnych kul. W modelu tym każdy atom tlenowca (tlenu, siarki, selenu) dotyka sześciu innych atomów tlenowca leżących w płaszczyźnie, która jest równoległa do warstwy x-M-x (x-atom tlenowca, M - atom metalu). Promień atomu tlenowca jest równy l/2ao (rys. 4.25). Atomy tlenowca leżą nad atomami warstwy x-M-x.
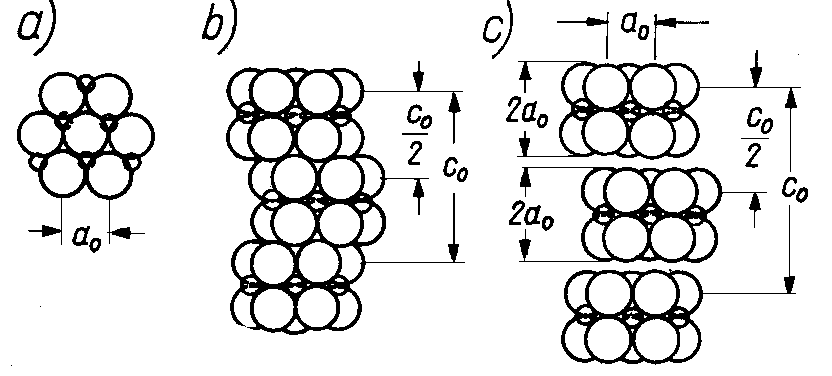
Rys. 4.25. Wpływ efektu upakowania na możliwości poślizgu:
- widok z góry,
- widok z boku siatki krystalicznej o niskim stosunku wymiarów osiowych,
- widok z boku siatki o stosunku wymiarów osiowych większych od 2
Grubość warstwy x-M-x jest równa 2ao. Warstwy x-M-x odznaczają się gęstym upakowaniem atomów (rys. 4.25b). Przy tak gęstym upakowaniu względny ruch pomiędzy warstwami jest bardzo utrudniony. Przy działaniu siły stycznej może następować wzajemne wypychanie się warstw lub deformacja położenia atomów
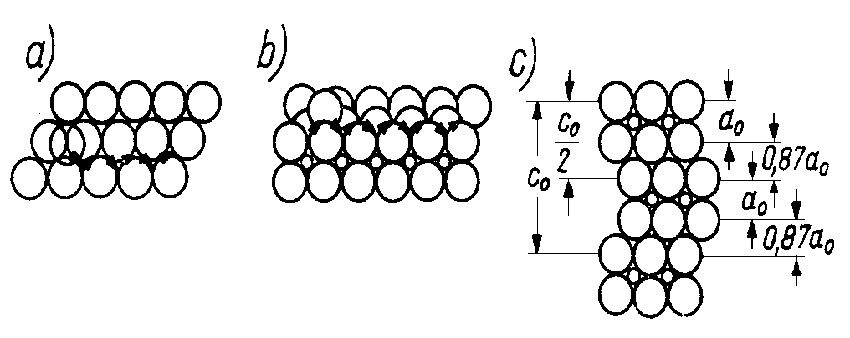
Rys. 4.26. Poślizg ruchem zygzakowatym:
- widok z góry,
- widok z boku,
- widok z boku siatki o najgęstszym upakowaniu
w siatce krystalicznej. Przy stopniowym zwiększaniu stosunku ao/co wzajemne przemieszczanie się warstw staje się coraz łatwiejsze i zupełne oddzielenie warstw, występujące przy stosunku ao/co równym 2, umożliwia swobodne przemieszczenie się warstw względem siebie w każdym kierunku równoległym do tych warstw (rys. 4.25). Znacznie mniejsze oddzielenie niż kompletne jest potrzebne, gdy poślizg ma nastąpić w jakimś określonym uprzywilejowanym kierunku. Jeżeli jest możliwy poślizg przy ruchu zygzakowatym po przylegającej warstwie, wówczas minimalna wartość stosunku ao/co wynosi 1,87 (rys. 4.26). Jeżeli poślizg ma przebiegać po linii prostej (rys. 4.27), wówczas stosunek ao/co musi wynosić minimum 1,90.
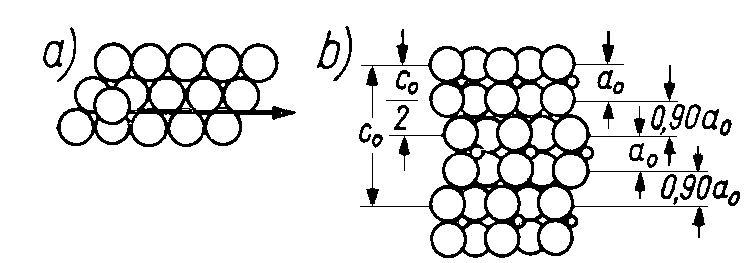
Rys. 4.27. Poślizg po linii prostej:
- widok z góry,
- widok z boku siatki o możliwym najgęstszym upakowaniu dla poślizgu po linii prostej
Wartości wyznaczone na podstawie modelu, w którym siatka krystaliczna jest złożona ze sztywnych kul, są zgodne z wartościami wyznaczonymi eksperymentalnie. Najniższa wartość stosunku ao/co, przy której występował poślizg z małymi oporami w badaniach kryształów tlenowców, wynosiła 1,93. W przypadku substancji krystalizujących w strukturach piramid trygonalnych wystarcza, aby stosunek ten wynosił 1,93, w przypadku stopów (stałych roztworów) dla osiągnięcia dobrego poślizgu jest konieczna wartość 1,96 (dla MoS2 wynosi ona 1,93, dla WS2 - 1,96).
Jak widać z powyższych rozważań przydatność substancji krystalizujących warstwowo zależy od przestrzennego ułożenia atomów wewnątrz siatki i dobre własności smarne osiąga się tylko przy odpowiednich wartościach stosunku ao/co. Dużą rolę w kształtowaniu własności smarnych mogą odgrywać również zanieczyszczenia, które mogą spowodować, że ta sama substancja może mieć dobre lub złe własności smarne. Dużą rolę odgrywają również zjawiska adsorpcji par i gazów na kryształach o budowie warstwowej. Bardzo duże zmiany wartości współczynnika tarcia w zależności od zjawisk adsorpcyjnych występują zwłaszcza w przypadku grafitu i dwusiarczku molibdenu. Przydatność substancji o budowie warstwowej jako smaru stałego zależy również od jej przyczepności do podłoża metalicznego.
Jamison i Cosgrove (26) stwierdzili, że występują następujące przypadki:
- substancja nie wykazuje przyczepności do podłoża metalicznego, które ma smarować,
- substancja jest przyczepna do podłoża metalicznego, ale nie jest odporna na działanie sił występujących przy poślizgu, nawet pod niewielkim obciążeniem i warstewka naniesiona na podłoże zostaje usunięta,
- substancja jest przyczepna do podłoża i jest odporna na działanie sił stycznych przy poślizgu, nawet przy znacznym obciążeniu.
Tylko trzecia grupa może mieć przydatność jako stałe substancje smarujące. Małej przyczepności do podłoża metalicznego można zapobiec przez dodawanie i zawieszanie smaru stałego w substancji wiążącej, którą jest zazwyczaj utwardzalna żywica syntetyczna lub lakier. Mieszaninę taką nanosi się na podłoże, które ma być smarowane. Żywica lub lakier związują poszczególne cząstki smaru stałego ze sobą i z podłożem; cząstki smaru zmniejszają wartość współczynnika tarcia. W przypadku niektórych smarów stałych jest możliwe ich osadzanie elektrochemiczne na podłożu (39) lub też wytwarzanie związku smarnego bezpośrednio w skojarzeniu trącym (np. wytwarzanie MoS2 in situ).
4.2.2. Rodzaje smarów stałych i teorie ich oddziaływania
Substancje stałe o małych oporach tarcia wewnętrznego, wykorzystywane jako smary stałe, można podzielić na dwie zasadnicze grupy:
- Substancje o małej wewnętrznej spójności:
- mydła, woski, stałe tłuszcze roślinne i zwierzęce,
- miękkie polimery,
- miękkie metale (ind, ołów, cyna, srebro, cynk, miedź), zazwyczaj
w postaci sproszkowanej.
- Substancje o spójności anizotropowej (o budowie warstwowej):
- grafit,
- siarczki metali przejściowych (np. MoS2, WS2, TiS2),
- selenki metali przejściowych (np. MoSe2, WSe2),
- tlenki (np. SbO3),
- wodorotlenki (np. Cd(OH)2, Mo(OH)2),
- halogenki (np. PbJ2, CdJ, AgJ, CdCl2 itd.),
- azotki (np. azotek boru BN),
- siarczany (np. Ag2SO4).
- Niektórzy autorzy są skłonni zaliczać w skład smarów stałych również powłoki ochronne wytwarzane na elementach maszyn i urządzeń, w procesach fosforowania, nasiarczania, cyjanonasiarczania itp. Mimo dużej różnorodności smarów stałych największe zastosowanie praktyczne znalazł graf it i dwusiarczek molibdenu. Nie należy się dziwić, że istniejące teorie tarcia smarów stałych wyjaśniają przede wszystkim ich zachowanie w skojarzeniach trących. W wyjaśnieniach proponowanych przez różnych autorów główną rolę przypisuje się albo czynnikom strukturalnym, albo też czynnikom związanym z procesami sorpcyjnymi. W odniesieniu do grafitu już Bragg (44) w swojej książce wydanej w 1928 r. sugeruje, że własności smarujące tej substancji są związane z charakterystyczną warstwową budową siatki krystalicznej, gdzie atomy węgla ułożone w warstwie są związane pomiędzy sobą silnymi wiązaniami, a pomiędzy atomami węgla znajdującymi się w sąsiednich warstwach działają znacznie mniejsze siły. Sugestie Bragga dały początek tzw. strukturalnej teorii tarcia smarów stałych.
- Wielu autorów uważa (41), (45), (46), że takie wyjaśnienia są nadmiernym uproszczeniem zagadnienia. Co prawda przyjmuje się, że energia wiązania atomów węgla znajdujących się w sąsiednich warstwach w graficie wynosi 4,18 do 83,72 J/mol (l do 20 kcal/mol), a pomiędzy atomami węgla ułożonymi w warstwie wynosi około 335 J/mol (80 kcal/mol), to jednak energia wiązań międzywarstwowych jest wielkością addytywną i suma energii tych wiązań może osiągać znaczne wartości. Wspomniani autorzy zasadniczą rolę w łatwym poślizgu między warstwami upatrują głównie w defektach struktury. Jako argument przeciw takiemu wyjaśnieniu własności smarujących materiałów o budowie warstwowej przedstawia się fakt, że kilka takich materiałów, jak np. mika, talk, dwusiarczek tytanu, pomimo że mają typową warstwową budowę, charakteryzują się wysokimi wartościami współczynnika tarcia. Dodatkowym argumentem jest fakt, że grafit oraz azotek boru w atmosferze powietrza, stanowiącej środowisko tarcia, wykazują małe wartości współczynników tarcia, podczas gdy w próżni wartość współczynnika tarcia ulega znacznemu zwiększeniu (35). W przypadku niektórych substancji występuje odwrotne zjawisko, tj. zmniejszenie wartości współczynnika tarcia w próżni (rys. 4.28, tabl. 4.12).
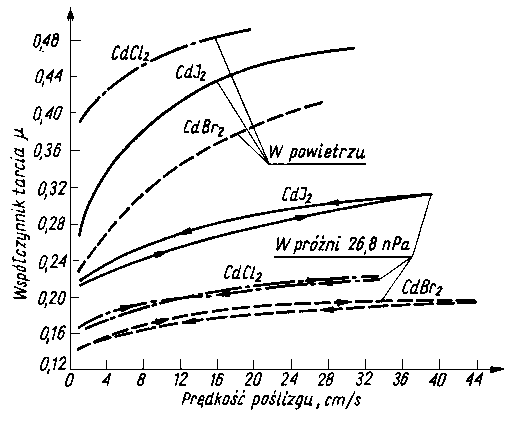
Rys. 4.28. Współczynnik tarcia halogenków kadmu w powietrzu i w próżni
- Na podstawie powyższych rozważań można przypuszczać, że w wielu przypadkach dużą rolę w procesach tarcia smarów stałych odgrywają zjawiska sorpcyjne, bowiem w środowisku powietrza przebiega sorpcja gazów i pary wodnej na powierzchniach smarów stałych, natomiast w próżni tego rodzaju procesy nie zachodzą. Pod uwagę należy brać również procesy utleniania.
- Omówione zjawiska dały podstawę do zaproponowania przyjęcia tzw. adsorpcyjnej teorii tarcia smarów stałych. W teorii tej nie neguje się wpływu czynników strukturalnych, niemniej jednak zasadniczą rolę przypisuje się procesom adsorpcji i utleniania.
- R. H. Savage (48) stwierdził, że w przypadku grafitu występują zmiany wartości współczynnika tarcia w powietrzu, tlenie i próżni. Zaproponował on teoretyczne wyjaśnienie tego zjawiska. Stwierdził on ponadto, że współczynnik tarcia grafitu zostaje wydatnie obniżony (w porównaniu z wartością w próżni) przy kontakcie kryształów grafitu z łatwo kondensującymi się parami, jak np. amoniaku, benzenu czy n-heksanu, natomiast efektu takiego nie stwierdza się przy kontakcie z atmosferą azotu czy wodoru. Savage uważa, że aby grafit mógł spełniać rolę smaru stałego, musi nastąpić zmniejszenie energii powierzchniowej w wyniku adsorpcji. Zwłaszcza ważne jest zmniejszenie dużej energii powierzchniowej krawędzi i naroży kryształów, których addytywne oddziaływanie powoduje wystąpienie dużej wartości współczynnika tarcia oraz silnych procesów zużycia. Nieaktywność wodoru tłumaczy Savage małą zdolnością tego gazu do pokrywania powierzchni grafitu (dla wodoru 1 ¸ 4%, a dla tlenu 20 - 25% powierzchni). Mała aktywność została stwierdzona również w przypadku azotu.
Tablica 4.12.
Wartości współczynników tarcia niektórych smarów stałych
w próżni i w powietrzu (47)
Ciało stałe Powietrze Próżnia Ciśnienie, µPa
Grafit naturalny 0,19 0,44 0,798
Grafit pirolityczny 0,18 0,50 0,266
- BN
0,25 0,70 0,266
- MoSz
0,18 0,07 0,266
- WSz
0,17 0,13 0,399
- BiJ3
0,34 0,39 66,50
- LiOH
0,37 0,21 0,266
- NiJ2
0,48 0,44 26,60
- CdCl2
0,35 0,16 0,266
- CdJ2
0,24 0,18 0,266
- CdBr2
0,22 0,15 0,266
- SnS2
0,40 1,0 0,266
Ftalocjanina 0,35 0,33 133,30
- Podobne rezultaty otrzymał w swoich badaniach Rowe (35). Badał on zachowanie się grafitu w wysokiej próżni, w atmosferze tlenu, pary wodnej i innych łatwo kondensujących się par. Stwierdził, że wrażliwość grafitu na sorpcję pary wodnej w dużym stopniu zależy od rodzaju zastosowanego grafitu.
- Do ciekawych rezultatów doszli w swoich pracach Campbell i Kozak (50). Badali oni tarcie pręta grafitowego o obracającą się tarczę miedzianą. Stwierdzili, że bardzo dużą rolę odgrywa chropowatość i twardość tarczy. W przypadku polerowanej tarczy miedzianej bardzo małe zużycie grafitu występuje nawet w atmosferze suchego azotu. Podobny efekt uzyskuje się, gdy kryształy w pręcie grafitowym są zorientowane równolegle do powierzchni tarczy. W przypadku zwiększenia chropowatości powierzchni tarczy i przy prostopadłej do powierzchni tarcia orientacji kryształów grafitu, małe zużycie uzyskuje się jedynie w obecności tlenu, pary wodnej i innych łatwo kondensujących się par.
- Cannon (51) stwierdza, że dużą rolę odgrywa długość łańcuchów zaadsorbowanych na powierzchni cząstek sorbatu. Badano również wpływ temperatury na wartość współczynnika tarcia grafitu. Stwierdzono, że powyżej 50°C wartość jego wzrasta od 0,1 do 0,4. Wzrost ten przypisuje się desorpcji pewnej ilości pary wodnej oraz procesom utleniania powierzchniowego.
- Zwolennicy adsorpcyjnej teorii tarcia smarów stałych zwracają ponadto uwagę na procesy adhezyjne. Twierdzą oni, że przy tarciu smarów stałych znacznie ważniejszą cechą, niż budowa warstwowa smaru, jest jego zdolność do adhezji, na powierzchni smarowanego metalu. Dobre smarowanie uzyskuje się wówczas, gdy związanie adhezyjne z powierzchnią metalu jest silniejsze niż siły wiązań pomiędzy warstwami smaru stałego, co umożliwia poślizg. Należy zaznaczyć, że obecnie zarówno teoria strukturalna, jak i adsorpcyjna, nie są w stanie wyjaśnić wszystkich zaobserwowanych zjawisk występujących przy tarciu smarów stałych.
4.2.3. Zastosowanie smarów stałych
Smary stałe są stosowane w tych przypadkach, gdy użycie innych rodzajów smarów jest niemożliwe lub niecelowe. Mogą być one stosowane przy bardzo dużych naciskach jednostkowych, przy których utrzymanie trwałej warstwy innych rodzajów smarów jest niemożliwe. Smary stałe odznaczają się również bardzo dużą odpornością na wysokie temperatury, przy których inne rodzaje smarów ulegają rozkładowi cieplnemu. Swoje własności smarne zachowują one nawet w niskich temperaturach, dlatego też znalazły one zastosowanie w wielu urządzeniach kriogenicznych. Ze względu na to, że są ciałami stałymi, mogą one być stosowane do procesów smarowania w wysokiej próżni, w której inne rodzaje smarów ulegają odparowaniu i nie mogą spełniać swoich funkcji smarnych. Zazwyczaj są one odporne na silne oddziaływanie radiacyjne, co pozwala na wykorzystanie ich w technice jądrowej, natomiast duża odporność na oddziaływanie agresywnego środowiska (kwasów, zasad i innych agresywnych chemikalii) umożliwia ich stosowanie w szerokim zakresie do smarowania różnego rodzaju elementów i urządzeń w przemyśle chemicznym.
- Duża trwałość smarów stałych pozwala na stosowanie ich w tych przypadkach, gdy jest konieczne jednorazowe nasmarowanie współpracujących elementów na cały okres pracy maszyny. Cecha ta ma również duże znaczenie przy trudnym lub niemożliwym w czasie eksploatacji dostępie do smarowanego skojarzenia trącego. Spośród wielu substancji stałych, odznaczających się zdolnością do zmniejszania wartości współczynnika tarcia, jako smary stałe mogą być stosowane tylko te substancje, które nie są higroskopijne czy rozpuszczalne w wodzie, jak również nie odznaczają się korozyjnością w stosunku do tworzyw, z których są wykonane smarowane elementy.
4.2.3.1. Grafit i dwusiarczek molibdenu
Najczęściej stosowanymi smarami stałymi są grafit i dwusiarczek molibdenu. Do smarowania w szczególnie wysokich temperaturach coraz częściej stosuje się również dwusiarczek wolframu. Grafit i dwusiarczek molibdenu (MoS2) są ciałami krystalicznymi (budowa warstwowa, heksagonalna, kolor szaro-czarny z metalicznym połyskiem). Twardość wg skali Mohsa grafitu wynosi 0,5 ¸ 1,5, dwusiarczku molibdenu-1-2. Gęstość grafitu wynosi 1,98 ¸ 2,38 kg/m³, MoS2 - 4,7 ¸ 4,8 kg/m³. Grafit jest termicznie trwały w próżni do temperatury 3500°C, w atmosferze powietrza do 550°C. W atmosferze tlenu utlenia się podobnie jak MoS2już w temperaturze pokojowej. Dwusiarczek molibdenu odznacza się mniejszą odpornością cieplną niż grafit (w próżni do 1200°C, w argonie do 1343°C, w atmosferze powietrza do 450°C). Utlenianie jest procesem działającym destrukcyjnie na grafit i dwusiarczek molibdenu. Szybkość utleniania zależy od wartości temperatury, w której przebiega proces, od stężenia tlenu, od czasu oddziaływania tlenu oraz od powierzchni kontaktu, a więc od wielkości ziarn kryształów. Dwusiarczek molibdenu w wyniku utleniania przechodzi w trójtlenek molibdenu (MoO3), który nie ma własności smaru stałego i pogarsza proces smarowania. Obszerne badania intensywności utleniania MoS2 w różnych temperaturach przeprowadził Vineal (56) (tabl. 4.13).
Tablica 4.13.
Intensywność utlenienia dwusiarczku molibdenu w różnych temperaturach (wg Vineala)
Temperatura °C Czas ogrzewania, h
1 3 6 18 24 48 72 96
zawartość procentowa trójtlenku molibdenu
250 < 0,1 < 0,1 < 0,1 < 0,1 0,2 0,4 0,5 0,6
300 1,0 3,2 5,0 15,0 19,5 24,3 26,0 27,3
400 2,9 12,1 16,0 22,5 24,5 27,9 27,0 46,0
500 57,2 74,5 79,3 80,2 84,7 94,7 96,0 96,5
- Obydwa podstawowe smary stałe mogą być otrzymywane ze źródeł naturalnych (ze złóż kopalnych - kopalny grafit i molibdenit) lub syntetycznie. Grafit syntetyczny otrzymuje się przez uwęglanie i grafityzację w wysokich temperaturach substancji bogatych w węgiel (np. węglowodorów), a syntetyczny dwusiarczek molibdenu przez zastosowanie wielu różnych metod, jak np. :
- z pierwiastków Mo+2S ® MoS2, przez prażenie w rurze żelaznej,
- z trójtlenku molibdenu w obecności siarkowodoru
MoO3 + 3H2S ® MoS2 + 3H2O + S
- z molibdenianu amonu
(NH4)2MoO4 + 2H2S ® (NH4)2MoS2 + 4H2O
(NH4)2MoS2+2H2 ® MoS2 MoS2 + H2S + (NH4)2S
- z molibdenianu amonu przez stapianie z siarką i za pomocą wielu innych metod.
- W Polsce produkcję syntetycznego dwusiarczku molibdenu opracowali Maciaszek (58) i Haś (57).
- Oddzielnym zagadnieniem jest sposób nanoszenia wspomnianych smarów stałych na powierzchnie smarowane. Smary te mogą być nanoszone lub kontaktowane ze smarowanymi powierzchniami w postaci zwartej (brykiety, ołówki i szczotki smarujące) lub w postaci rozdrobnionej, tj. w postaci proszku. Proszki mogą być wcierane w smarowane powierzchnie lub też nanoszone w postaci zawiesin i aerosoli. Stosuje się zawiesiny wodne smarów stałych lub w łatwo lotnych rozpuszczalnikach. Sporządzane są również różnego rodzaju pasty. Stosunkowo duże ilości smarów stałych są zawieszane w olejach smarnych oraz w smarach plastycznych. Dla zwiększenia przyczepności do podłoża sproszkowane smary stałe są zawieszane w lakierach, którymi maluje się smarowane powierzchnie, lub też w szkle wodnym (roztwór krzemianu sodu), które powodują powiązanie z podłożem. W szerokim zakresie, jako materiały wiążące, wykorzystywane są różnego rodzaju żywice syntetyczne. Grafit i dwusiarczek molibdenu wprowadza się również do łożysk ze spieków oraz do plastycznych tworzyw niskotarciowych.
4.2.3.2. Miękkie metale
Miękkie metale znalazły zastosowanie w trybotechnice w postaci sproszkowanej, jak również w postaci zwartej, jako niskotarciowe materiały łożyskowe. Największe zastosowanie w postaci sproszkowanej znalazły proszki miedzi, glinu, cyny, ołowiu, a w niektórych przypadkach i srebra. Stosowane są również proszki stopów miękkich metali.
- Miękkie metale są ponadto nanoszone w postaci bardzo cienkich powłok (grubość ok. 10 -6 µm na twardym podłożu, w celu zabezpieczenia tego podłoża przed zatarciami w przypadku przerwania smarnej warstwy olejowej. Stanowią więc one dodatkowe zabezpieczenie elementów przed zatarciem. Do wytwarzania miękkich powłok na twardym podłożu najczęściej są stosowane: ind, ołów, gal i kadm. Szczególnie dobre rezultaty otrzymuje się w przypadku powłok z indu.
4.2.3.3. Polimery niskotarciowe
Spośród polimerów odznaczających się małą wartością współczynnika tarcia największe zastosowanie znalazły: teflon (policzterofluoroetylen) oraz poliamidy (nylon).
- Teflon, jako polimer niskotarciowy, ma zarówno wiele zalet, jak i wad. Z zalet należy wymienić: najmniejszą wartość współczynnika tarcia z dotychczas znanych materiałów (bez smarowania), brak zjawiska „stick-slip" podczas tarcia, obojętność chemiczna, niewrażliwość na działanie agresywnego środowiska oraz możliwość pracy w stosunkowo wysokich temperaturach (do 300°C). Do wad, w poważnym stopniu ograniczających jego zastosowania, należą: mała przewodność cieplna, duża rozszerzalność termiczna, „płynięcie na zimno" pod wpływem obciążeń ściskających oraz mała odporność na zużycie. Dla wyeliminowania wad teflonu stosuje się go w postaci wzmocnionej włóknem szklanym, metalowym, azbestowym lub innym oraz tworzy się mieszaniny z polimerami o dużej wytrzymałości mechanicznej, często z zastosowaniem wypełniacza w postaci proszku grafitowego czy też MoS2. Praktykowane jest również nanoszenie cienkich powłok z teflonu na metalowe podłoża lub też wypełnianie materiałów porowatych teflonem (np. łożyska wg patentu firmy Glacier). Teflon w postaci sproszkowanej może być również nanoszony na powierzchnie smarowane w postaci zawiesin (np. past, w smarach plastycznych itp.). Ten sposób wykorzystania teflonu w technice smarowniczej zapewnia uzyskanie bardzo dobrych efektów.
- Sproszkowane poliamidy, jako materiały niskotarciowe, nie zapewniają takich efektów jak teflon, ale w postaci zwartej są w szerokim zakresie stosowane jako materiał na elementy ślizgowe łożysk pracujących w umiarkowanych temperaturach i przy niewysokich obciążeniach. W szerokim zakresie są również stosowane poliamidy z wypełniaczem grafitowym lub z MoS2.
| Mt = N f = N µobl d | (4.6) |
| P ≥ T ≥ N µobl | (4.7) |
Tablica 4.3. | Typ łożyska | Współczynnik tarcia µr | Kulkowe zwykle przy obciążeniu promieniowym | 0,002 | Kulkowe zwykłe przy obciążeniu osiowym | 0,004 | Kulkowe wahliwe | 0,001 5 | Walcowe z wałeczkami walcowymi krótkimi | 0,003 | Walcowe z wałeczkami walcowymi długimi | 0,006 | Igiełkowe | 0,008 | Baryłkowe | 0,004 | Kulkowe wzdłużne | 0,003 | Stożkowe przy obciążeniu promieniowym | 0,008 | Stożkowe przy obciążeniu osiowym | 0,020 | |
4.1.5. Hipotezy tarcia suchego ciał stałych
Mimo długotrwałych badań występującego powszechnie w przyrodzie i technice zjawiska tarcia zewnętrznego ciał stałych, nieodłącznie związanego z wszelkiego rodzaju ruchem w ośrodku materialnym, nie powstała dotychczas ogólna teoria tarcia suchego.
Tablica 4.4. | Grupy teorii | Nazwa teorii | Podstawowe wzory | Cechy teorii | Mechaniczne | Amontonsa Coulomba | T = µ N T = A + µ N | Teoria pokonywania nierówności powierzchni; nie zależy od N, A nie zależy od rzeczywistego pola styku | Bowdena | T = S tpl + Sw pw | Teoria tworzenia i niszczenia szczepień; opory tarcia są sumą oporów ścinania nierówności szczepionych i przepychania odkształconego materiału | Molekularne | Tomlinsona |
| Opory tarcia są oporami pokonywania przyciągania molekularnego | Dieriagina | T = µ(N+No) N = PoSr | Pokonywanie chropowatości molekularnej ; nie uwzględnia własności materiału | Molekularno-mechaniczna | Kragielskiego |
| Tarcie = suma oporów pokonywania szczepień, chropowatości i przyciągania molekularnego; a= f(ścinania); b = f( powiększania oporu na ścinanie od N). |
| |||||||||||||||
określonym przez zespół różnych zjawisk zależnych od parametrów i warunków tarcia. Począwszy od XV wieku (Leonardo da Vinci) wielu wybitnych uczonych przeprowadzało badania zagadnień tarcia i formułowało zależności, z których można obliczyć wartość współczynnika tarcia.
4.1.5.1. Mechaniczne teorie tarcia suchego
Mechaniczne teorie tarcia suchego były najwcześniej opracowane. Teorie te objaśniają opór tarcia jako czynnik przeciwdziałający wykonywanej pracy, zużywanej na: unoszenie ślizgającego się elementu maszyny po nierównościach powierzchni drugiego elementu, na ścinanie nierówności i połączeń tarciowych oraz na pokonanie oporów wywołanych odkształceniami sprężystymi i plastycznymi w mikroobszarach styku.| T = µ N | (4.8) |
|
| Rys. 4.3. Model oddziaływania obszarów styku trących powierzchni |
Amontons stwierdził, że „siła tarcia znajduje się w złożonej zależności od nacisku jednostkowego, czasu i prędkości ślizgania przy tarciu ruchowym". Prawo Amontonsa zakłada, że trące ciała są idealnie sztywne.
| T = µ N + A | (4.9) |
W równości tej składnik A jest poprawką do wzoru Amontonsa i przedstawia część siły tarcia zależnej od molekularnego oddziaływania (sczepności) powierzchni trących.
-
Przy takich założeniach Bowden podzielił wszystkie przypadki tarcia ślizgowego na trzy rodzaje:
- ślizganie się metalu twardego po metalu twardym,
- ślizganie się metalu miękkiego po metalu twardym,
- ślizganie się metali o jednakowej twardości.
|
Rys. 4.4. Sczepianie się obszarów styku trących powierzchni; |
Rys. 4.5. Model Bowdena do obliczania i interpretacji siły tarcia suchego |
| S = |
N Re | (4.10) |
| T = Pś + Pw | (4.11) |
| Pś = Sw tpt | (4.12) |
gdzie: tpt - wytrzymałość na ścinanie metalicznych połączeń w kierunku stycznym do płaszczyzny tarcia, S - jak we wzorze (4.10).
| Pw = Sw pw | (4.13) |
| Sw = |
d ³ 12r | (4.14) |
| T = S tpt + Swpw | (4.15) |
| T = l d tpt + |
d ³ 12r | pw | (4.16) |
| T = |
d ³ 12r | pw | (4.17) |
Tablica 4.5. | Metal | Doświadczalne wartości wytrzymałości na ścinanie metalicznych połączeń tpt MPa | Wytrzymałość na ścinanie metalu przy próbie statycznej t MPa | Ind | 3,185 | 2,156 | Ołów | 15,680 | 7,350 | Miedź | 27,400 | 156,800 | Stal | l 372,000 | 882,000 |
| |||||
| T = S tpt | (4.18) |
|
Rys. 4.6. Model Epifanowa do obliczania składowej siły tarcia suchego |
Rys. 4.7. Zagłębianie i przesuwanie narostu metalu podczas ruchu elementu |
| T = t Sc | (4.19) |
| t = to + k sp | (4.20) |
| T = to Sc + k Sc sp | (4.21) |
a przyjmując
| sp = |
N Sc | (4.22) |
otrzymamy
| T = to Sc + k Sc sp | (4.23) |
| T = N | ( |
to spl | k | ) | (4.23) |
| µ = | to spl | k |
Tablica 4.6. | Metoda określenia wytrzymatości na ścięcie | Aluminium | Miedź | Żelazo | Stal 417 | Stop 437 | Stop WT-2 | Mosiądz | Badanie tarcia | 8,3 | 29,2 | - | - | - | - | - | 7,4 | 24,5 | 56 | 88 | 106 | 103 | 42 | Próby statyczne | 8,0 | 26,5 | 51 | 81 | 108 | 92 | 40 |
W pierwszej rubryce poziomej są podane wartości obliczone z pól śladów ścięcia, w następnej obliczone z pola koła o średnicy równej szerokości bruzdy tarcia.
| ||||||||||||||
|
|
Rys. 4.8. Narost tworzący się przed czołową płaszczyzną ślizgacza;
|
4.1.5.2. Adhezyjne teorie tarcia suchego
Osiągnięcia technologiczne uzyskane w obróbce powierzchniowej, dzięki której uzyskano znaczną gładkość powierzchni, nie doprowadziły do wyeliminowania przy ślizganiu dużych oporów tarcia. Omówione więc dotychczas mechaniczne teorie tarcia suchego nie wyjaśniły przyczyny tego zjawiska. Wysunięto więc hipotezę, że opór tarcia jest spowodowany oddziaływaniem pól sił atomów, cząsteczek lub jonów, a więc adhezją.| N = | S | Pi |
|
Rys. 4.9. Model Tomlinsona działania sił rniędzycząsteczkowych |
Rys. 4.10. Model Tomlinsona równoważenia obciążenia normalnego siłami odpychającymi |
Podczas ślizgania po sobie dwóch powierzchni zachodzi ciągła zamiana par cząsteczek znajdujących się w zbliżeniu i równoważących obciążenie normalne. Przy przesuwaniu jednego ciała względem drugiego siły odpychania przechodzą w siły przyciągania, a te ostatnie przeciwstawiają się ślizganiu.
| N = n P | (4.25) |
| k = n |
x l | (4.26) |
| k = n b |
x l |
| LT = µ n P x | (4.27) |
a ponieważ
| k E = LT | (4.28) |
zatem
| µ = |
k E n p x | = |
E l P | (4.29) |
| n = f( Sr ) | (4.30) |
gdzie Sr - rzeczywista powierzchnia styku zależna od obciążenia normalnego N.
| µ = |
E b l N | f( Sr ) | (4.31) |
| µ = 0,18 • 10 8 ( Ak + Ap ) 2/3 | (4.32) |
gdzie Ak i Ap - moduły zależne od własności mechanicznych materiału.
|
Rys. 4.11. Model mechaniczny Deriagina oddziaływania atomów w obszarze tarcia |
Rys. 4.12. Rozkład sił wg Deriagina podczas tarcia w obszarze styku atomów |
|
F N | = tg a | (4.33) |
| F = N tg amax = µ N | (4.34) |
gdzie µ = tg amax - statyczny współczynnik tarcia.
| T = (N + No) µ | (4.35) |
| µo = |
T N | = µ | ( | 1 + |
No N | ) | (4.36) |
| T = µ (N + po Sr ) | (4.37) |
| Sr = |
N p 'o | (4.38) |
| To = µ | ( | 1 + |
po p 'o | ) | N | (4.39) |
4.1.5.3. Adhezyjno-mechaniczna teoria tarcia suchego
W celu uwzględnienia w teorii tarcia zgodności zasad teoretycznych z wynikami badań sformułowano mechaniczno-adhezyjną teorię tarcia suchego. Autorem tej teorii jest Kragielski. W teorii tej Kragielski siły adhezji utożsamia z siłami oddziaływania molekularnego. Założył on, że siły tarcia występują w dwu postaciach. A mianowicie na rzeczywistych trących powierzchniach styku występuje mechaniczny opór ruchu, spowodowany zaczepieniem o siebie nierówności powierzchni, oraz opór spowodowany oddziaływaniem sił molekularnych, uwarunkowany wzajemnym przyciąganiem atomów lub cząsteczek trących ciał.| n | ||||
| To = Sr | ∑ | Fji | (4.40) | |
| 1 | ||||
| Fj mol = a1 + b1 q | (4.41) |
| Fj mech = a2 + b2 q | (4.42) |
| Fj = Fj mol + Fj mech = a + b q | (4.43) |
| Tmol = Fj mol Sr 1 | (4.44) |
a składowa siły tarcia wywołana oddziaływaniem mechanicznym
| Tmech = Fj mechl Sr 2 | (4.45) |
| Sr = Sr 1 + Sr 2 | (4.46) |
| T = a Sr + b N | (4.46) |
Współczynnik tarcia µ można określić z zależności
| µ = |
T N | = |
a Sr N | + b | (4.48) |
| a = |
n a2 + a1 n +1 | ; b = |
n b2 + b1 n +1 | (4.49) |
gdzie: a1, a2 oraz b1, b2 są objaśnione ze wzorami (4.41) i (4.42).
|
| (4.50) |
| µ = |
a spl | + b | (4.51) |
|
| (4.52) |
|
Rys. 4.13. Model rozkładu naprężeń w obszarze styku dwóch ciał chropowatych |
Rys. 4.14. Model falowego odkształcenia metalu w obszarze zagłębionego występu nierówności |
Proces takiego odkształcenia materiału polega na przemieszczaniu fali materiału przed występem oraz odsuwanie materiału na boki z bruzdy zakreślonej występem nierówności.
4.1.6. Zjawiska w strefie kontaktu ciało stałe-ciało stałe
4.1.6.1. Adhezja
Adhezja jest zjawiskiem powierzchniowym, polegającym na sczepieniu się powierzchni stykających się ciał wskutek oddziaływania między nimi pola sił. Pole sił wytwarzane przez ładunki atomów (jonów, cząsteczek), z których jest zbudowana warstwa wierzchnia ciał, maleje wykładniczo wraz ze wzrostem odległości od powierzchni. Praktycznie oddziaływanie powierzchniowe typu Van der Waalsa zanika powyżej odległości 1-2 nm, dlatego dla zaistnienia sczepienia adhezyjnego jest konieczne odpowiednie zbliżenie powierzchni. Dla uzyskania dużej rzeczywistej powierzchni styku stosuje się polerowanie.4.1.6.2. Zjawiska adhezyjno-dekohezyjne
Przy wzajemnym kontakcie dwóch ciał stałych występuje styk powierzchniowy. Rozróżnia się kontakt ciał nieruchomych i kontakt ciał przemieszczających się względem siebie. Przy kontakcie idealnie gładkich powierzchni, rzeczywista powierzchnia styku byłaby równa nominalnej powierzchni styku. Ponieważ realna powierzchnia styku ma mnóstwo nierówności i styk między powierzchniami występuje jedynie na nierównościach, dlatego też rzeczywista powierzchnia styku stanowi jedynie niewielki ułamek powierzchni nominalnej. Jak już wspomniano na powierzchni ciała występuje pole sił, którego oddziaływanie na znajdujące się w jego zasięgu ciała powoduje przeciąganie - adhezję. Powiązanie adhezyjne jest tym silniejsze, im silniejsze jest oddziaływanie elektrostatyczne i elektrodynamiczne pomiędzy zetkniętymi ze sobą ciałami. Wiąże się to ściśle z budową ciał, a więc i ich powierzchni. Siły adhezyjne mają charakter addytywny, dlatego też im większa jest rzeczywista powierzchnia styku, tym silniejsze występuje sczepienie adhezyjne. Oddziaływanie adhezyjne maleje bardzo gwałtownie (wykładniczo) wraz ze wzrostem odległości między kontaktującymi się ciałami.| Tt = Tadh + Tkoh |
Jest to najogólniejsze prawo trybologiczne. W zależności od rodzaju i warunków tarcia poszczególne człony mogą wzrastać lub maleć, albo nawet zupełnie zanikać. Tak np. przy tarciu wewnętrznym cieczy człon adhezyjny jest zbliżony do zera, natomiast przy tarciu zewnętrznym idealnie gładkich powierzchni człon kohezyjny byłby równy zeru. Oprócz wymienionych tu skrajnych przypadków, w których jeden z członów zanika, istnieje wiele przypadków pośrednich, w których obydwa człony -adhezyjny i kohezyjny-mają znaczną wartość. Powyższe ujęcie ma duże znaczenie ze względów pojęciowych, natomiast dokładne obliczenie wartości poszczególnych członów jest trudne. Istniejące wzory pozwalają na obliczanie zasadniczych elementów obu członów, a stopniowe poznawanie zjawisk trybologicznych umożliwi w przyszłości zwiększanie dokładności obliczeń.
|
|
| Rys. 4.15. Tryboelektryczny szereg polimerów |
tworzenia się ładunków elektrostatycznych. Zaznacza się jednak znaczny wpływ metalu, a zwłaszcza jego zdolność do odprowadzania ładunku. Należy jednak podkreślić, że możliwe jest znaczne oddziaływanie adhezyjne metal-polimer przy dużej gładkości powierzchni polimeru. Tak np. w laboratorium podczas badań przeprowadzonych przez autorów stwierdzono zużywanie się stali przy jej kontakcie z przeciwpróbką z metakrylanu metylu (plexiglas) o lustrzanie gładkiej powierzchni. Powierzchnia polimeru zostaje jednak zazwyczaj stosunkowo łatwo niszczona mechanicznie (rysowanie, bruzdowanie, deformacja) i w tym przypadku człon kohezyjny ma dużą wartość. Polimery wewnętrznie spójne, mające odporną warstwę powierzchniową, w przypadku jej znacznej gładkości powodują małe sumaryczne opory tarcia, gdyż zarówno człon adhezyjny, jak i kohezyjny mają małą wartość.
4.1.7. Tarcie monokryształów
Badanie tarcia monokryształów ma na celu przede wszystkim ustalenia podstawowych praw dotyczących tarcia ciał stałych. Umożliwia ono określenia uprzywilejowanych płaszczyzn i kierunków poślizgu, powiązanie oporów tarcia z gęstością upakowania atomowego i ułożenia płaszczyzn atomowych. Odpowiednio zaprogramowane badania umożliwiają również śledzenie wpływu defektów budowy na opory tarcia monokryształów oraz powiązania ich z energią powierzchni, współczynnikiem sprężystości i twardością. Przeprowadzone badania monokryształów czystych metali i ich stopów oraz ciał krystalicznych o budowie jonowej i kowalentnej pozwalają na dokonanie pewnych uogólnień. Dla tego celu szczególnie duże znaczenie mają prace H. D. Buckley'a (12), (13), (14), (15), na których w głównej mierze oparto przedstawione rozważania. Jak wiadomo płaszczyzny poślizgu w kryształach są zazwyczaj najgęściej upakowanymi płaszczyznami i pomiędzy tymi płaszczyznami występują w krysztale największe odstępy. Jeżeli w krysztale występuje poślizg, to zazwyczaj wzdłuż kierunku o najgęstszym upakowaniu, gdyż znacznie większe naprężenie ścinające jest potrzebne dla zainicjowania poślizgu płaszczyzn o mniejszej gęstości upakowania. Pewne zaburzenia od tej reguły występują w przypadku kryształów o więzi jonowej (np. soli kuchennej).|
|
|
|
| |
|
|
można wywołać odkształcenie plastyczne. Należy jednak zaznaczyć, że w takich warunkach zwiększa się również tendencja do występowania umocnień. Do badań tarcia monokryształów Buckley stosował urządzenie próżniowe przedstawione na rys. 4.17. Skojarzenie trące składało się z tarczy i ślizgacza wykonanego z monokryształu. Tarcza jest zamocowana na trzpieniu napędzanym dwudziestobiegunowym magnesem poprzez diafragmę. Magnes napędzający znajdował się na zewnątrz układu próżniowego. Ślizgacz umocowany był na ruchomym ramieniu przenoszącym zarówno obciążenie, jak i siłę tarcia. Dla usunięcia z powierzchni trących zaadsorbowanych substancji w układ próżniowy wmontowano działo elektronowe. Rezultaty badań zostaną przedstawione oddzielnie dla różnych typów struktur krystalicznych.
4.1.7.1. Kryształy o więzi metalicznej
Sieć ściennie centrowana układu regularnego. Dla metali krystalizujących w tej sieci stwierdzono anizotropowość oporów tarcia. Najmniejsze opory tarcia występują przy poślizgu na uprzywilejowanych kierunkach, tj. na płaszczyznach o największej gęstości atomowej. Jak widać z tabl. 4.7 zwiększenie gęstości atomowej powoduje
Tablica 4.7. | Płaszczyzna | Gęstość atomowa (liczba atomów/jednostkę powierzchni) | Odstęp pomiędzy płaszczyznami nm | Energia powierzchniowa
| Współczynnik tarcia | Powietrze 1000 hPa | Próżnia 1,33 mPa | (100) | 0,152 7 | 0,128 | 2 892 | 0,60 | > 40 | (110) | 0,108 0 | 0,180 5 | - | 40 | > 40 | (111) | 0,172 2 | 0,208 | 2499 | 21 | 21 | |||||||||
zmniejszenie energii powierzchniowej, zmniejszenie współczynnika tarcia oraz zwiększenie współczynnika sprężystości.
Tablica 4.8. | Typ struktury krystalicznej | Płaszczyzna poślizgu | Kierunek poślizgu | Liczba możliwych wariantów poślizgu | Przykłady | Kryształy o więzi metalicznej | Sieć ściennie centrowana układu regularnego | (111) | (110) | 12 | Cu, Ni, Al, Ag, Au | Sieć przestrzennie centrowana układu regularnego | (110) (112) (123) | (111) (111) (111) | 12 12 24 | Fe, W, Mo |
Sieć heksagonalna zwarta - poślizg na płaszczyźnie: | (0001) | (1120) | 3 | Co Be Re Cd Zn La Ti Zr Hf | Kryształy o więzi jonowej |
Sieć regularna (sól kuchenna) Sieć romboedryczna | (110) (0001) | (110) (1120) | 12 3 | MgO, LiF, NaCl, KBr, Al2O3 | Kryształy o więzi kowalencyjnej | Diament | (111) | (110) | 12 | Si, Ge, diament | ||||||||||||||||
do adhezji, powodują mniejsze opory tarcia oraz mniejsze zużycie niż metale układu regularnego. Wyraźnie mniejsza jest również skłonność do zacierania. Uwydatnia się u nich wyraźna anizotropowość własności, między innymi również własności tarciowych. Badania tarcia monokryształów metali krystalizujących w sieci heksagonalnej, jak kobaltu, berylu i renu (tabl. 4.9) wykazały, że najmniejsze opory tarcia występują przy poślizgu w płaszczyźnie podstawy (0001). W tym kierunku występuje również największa gęstość upakowania atomowego oraz najniższy współczynnik tarcia. Ze względu na to, że poślizg w płaszczyźnie podstawy jest możliwy w trzech kierunkach, współczynnik tarcia przyjmuje tę samą wartość, co przy kącie 60° ułożenia płaszczyzny podstawy monokryształu do powierzchni tarcia.
Tablica 4.9. | Płaszczyzna poślizgu | (0001) | (1010) | Kierunek poślizgu | (1120) | (1010) | (1120) | (0001) | Kobalt | 0,38 | - | 0,80 | - | Beryl | 0,48 | 0,51 | - | 0,70 | Ren | 0,28 | 0,38 | 0,50 | 0,80 | Tytan | 0,56 | - | 0,36 | - | |||||
|
|
| Rys. 4.18. Współczynnik tarcia dla monokryształu berylu trącego o polikrystaliczną tarczę berylową w próżni przy różnych kątach Q między płaszczyzną podstawy a płaszczyzną powierzchni (obciążenie 2,50 N prędkość poślizgu 0,13 mm/s, ciśnienie 1333 nPa bez podgrzewania zewnętrznego próbki) |
Jak widać z rysunku współczynnik tarcia ma największą wartość przy prostopadłym (kąt 90°) ułożeniu płaszczyzny podstawy monokryształu berylu do powierzchni tarcia. Interesujący jest fakt, że najmniejszej wartości współczynnika tarcia nie obserwowano przy równoległym ułożeniu płaszczyzny podstawy monokryształu do powierzchni tarcia, lecz przy kącie 135°. Dla tytanu występują mniejsze opory tarcia w płaszczyźnie pryzmatycznej (1010) niż w płaszczyźnie podstawy (0001), gdyż (w odróżnieniu od innych metali tej grupy) płaszczyzna podstawy nie jest płaszczyzną najgęściej upakowaną i większą gęstość upakowania ma płaszczyzna pryzmatyczna (1010).
4.1.7.2. Kryształy o więzi jonowej
Dla kryształów o więzi jonowej zachowanie w procesach trybologicznych jest uwarunkowane siłą oddziaływania jonowego między elektrododatnim jonem metalu i silnie elektroujemnym jonem chlorowca, tlenu itp. Kryształy tego typu mają bardzo ograniczone zastosowania trybologiczne, dlatego też badaniami obejmowane są dla uzyskania poglądu na ich zachowanie się podczas procesów tarcia różnych grup substancji krystalicznych. Stosunkowo najbardziej zainteresowano się krystaliczną budową tlenku glinu - szafiru, który stosowano jako materiał na łożyska w różnego rodzaju przyrządach precyzyjnych. Stwierdzono, że szafir wykazuje anizotropię zachowania się podczas tarcia zbliżoną do metali krystalizujących w układzie heksagonalnym. W badaniach tarcia szafiru stwierdzono, że przy ruchu postępowo-zwrotnym występują różne opory i intensywność zużywania jest zależna od kierunku ruchu(rys. 4.18, tabl. 4.10). Zjawisko to niektórzy uzasadniają budową kryształu szafiru podobną do rozsuniętej nieco talii kart, co powoduje różne zachowanie się w zależności od kierunku ruchu. Kryształy jonowe krystalizujące w układzie regularnym|
|
Rys. 4.19. Zależność intensywności zużywania kryształu szafiru od kąta Q i kierunku tarcia; |
Tablica 4.10. | Płaszczyzna poślizgu | Współczynnik tarcia | Kierunek poślizgu | (0001) | (1120) | 0,50 | (1010) | 0,96 | (1010) | (1120) | 0,93 | (0001) | 1,00 | 1) Warunki: obciążenie 10 N, v = 0,013 cm/s, temperatura 20°C, ciśnienie 13,3 nPa. | ||||
zachowują się zupełnie inaczej niż metale krystalizujące w tym samym układzie. Tak np. dla soli kuchennej płaszczyzną o największej gęstości atomowej jest płaszczyzna (100), a płaszczyzną o najłatwiejszym poślizgu - płaszczyzna (100). W przypadku Nad naprężenie ścinające jest w płaszczyźnie (100) cztery razy większe niż w płaszczyźnie (110), a dla LiF-sześć razy większe (w temperaturze 100°C). Zjawiska te wynikają z oddziaływań międzyjonowych.
4.1.7.3. Kryształy o więzi kowalentnej
Elementy budowy kryształów tego typu są powiązane pomiędzy sobą silnymi wiązaniami kowalentnymi. W przypadku diamentu występuje koordynacja tetraedryczna atomów węgla. Jak wykazały badania najmniejsze opory tarcia w przypadku diamentu występują w płaszczyźnie najgęstszego upakowania atomów (tabl. 4.11).
Tablica 4.11. | Płaszczyzna poślizgu | Kierunek poślizgu | Energia powierzchniowa kJ/m² | Współczynnik tarcia | przy 1000 hPa | przy 13,3 nPa | (100) | (100) | 7050 | 0,15 | - | (110) | - | 0,05 | - | (111) | -1) | 3500 | 0,05 | 0,9 | 1) Kierunek na płaszczyźnie (111) nie ma wpływu na współczynnik tarcia. | |||||||
4.2. Tarcie wewnętrzne ciał stałych. Smary stałe
4.2.1. Budowa i tarcie wewnętrzne smarów stałych
Tarcie zewnętrzne ciał stałych charakteryzuje się z reguły znacznymi oporami. W celu zmniejszenia oporów tarcia wprowadza się między współpracujące powierzchnie smar, tj. substancję o małych oporach tarcia wewnętrznego, przez co zastępuje się tarcie zewnętrzne (suche) ciał stałych tarciem wewnętrznym. Do smarowania stosuje się zazwyczaj smary ciekłe (oleje) lub smary plastyczne. Smary te mają jednak ograniczoną odporność mechaniczną i cieplną. Znacznie większą odporność mechaniczną i cieplną mają smary stałe. Nazwą tą określa się ciała stałe odznaczające się stosunkowo małymi siłami spójności i dające się w związku z tym łatwo odkształcać plastycznie. Mała spójność może charakteryzować albo całe ciało, albo też może ona występować tylko w pewnych uprzywilejowanych kierunkach poślizgu, a więc wtedy ciało pod względem spójności ma charakter anizotropowy. Anizotropowością|
| Rys. 4.20. Siatka krystaliczna grafitu |
|
|
|
sił spójności odznaczają się przede wszystkim ciała stałe o warstwowej budowie siatki krystalicznej.
|
|
|
|
(a)
|
(b)
|
|
|
w siatce krystalicznej. Przy stopniowym zwiększaniu stosunku ao/co wzajemne przemieszczanie się warstw staje się coraz łatwiejsze i zupełne oddzielenie warstw, występujące przy stosunku ao/co równym 2, umożliwia swobodne przemieszczenie się warstw względem siebie w każdym kierunku równoległym do tych warstw (rys. 4.25). Znacznie mniejsze oddzielenie niż kompletne jest potrzebne, gdy poślizg ma nastąpić w jakimś określonym uprzywilejowanym kierunku. Jeżeli jest możliwy poślizg przy ruchu zygzakowatym po przylegającej warstwie, wówczas minimalna wartość stosunku ao/co wynosi 1,87 (rys. 4.26). Jeżeli poślizg ma przebiegać po linii prostej (rys. 4.27), wówczas stosunek ao/co musi wynosić minimum 1,90.
|
-
Jamison i Cosgrove (26) stwierdzili, że występują następujące przypadki:
- substancja nie wykazuje przyczepności do podłoża metalicznego, które ma smarować,
- substancja jest przyczepna do podłoża metalicznego, ale nie jest odporna na działanie sił występujących przy poślizgu, nawet pod niewielkim obciążeniem i warstewka naniesiona na podłoże zostaje usunięta,
- substancja jest przyczepna do podłoża i jest odporna na działanie sił stycznych przy poślizgu, nawet przy znacznym obciążeniu.
4.2.2. Rodzaje smarów stałych i teorie ich oddziaływania
-
Substancje stałe o małych oporach tarcia wewnętrznego, wykorzystywane jako smary stałe, można podzielić na dwie zasadnicze grupy:
- Substancje o małej wewnętrznej spójności:
- mydła, woski, stałe tłuszcze roślinne i zwierzęce,
- miękkie polimery,
- miękkie metale (ind, ołów, cyna, srebro, cynk, miedź), zazwyczaj w postaci sproszkowanej.
- Substancje o spójności anizotropowej (o budowie warstwowej):
- grafit,
- siarczki metali przejściowych (np. MoS2, WS2, TiS2),
- selenki metali przejściowych (np. MoSe2, WSe2),
- tlenki (np. SbO3),
- wodorotlenki (np. Cd(OH)2, Mo(OH)2),
- halogenki (np. PbJ2, CdJ, AgJ, CdCl2 itd.),
- azotki (np. azotek boru BN),
- siarczany (np. Ag2SO4).
- Niektórzy autorzy są skłonni zaliczać w skład smarów stałych również powłoki ochronne wytwarzane na elementach maszyn i urządzeń, w procesach fosforowania, nasiarczania, cyjanonasiarczania itp. Mimo dużej różnorodności smarów stałych największe zastosowanie praktyczne znalazł graf it i dwusiarczek molibdenu. Nie należy się dziwić, że istniejące teorie tarcia smarów stałych wyjaśniają przede wszystkim ich zachowanie w skojarzeniach trących. W wyjaśnieniach proponowanych przez różnych autorów główną rolę przypisuje się albo czynnikom strukturalnym, albo też czynnikom związanym z procesami sorpcyjnymi. W odniesieniu do grafitu już Bragg (44) w swojej książce wydanej w 1928 r. sugeruje, że własności smarujące tej substancji są związane z charakterystyczną warstwową budową siatki krystalicznej, gdzie atomy węgla ułożone w warstwie są związane pomiędzy sobą silnymi wiązaniami, a pomiędzy atomami węgla znajdującymi się w sąsiednich warstwach działają znacznie mniejsze siły. Sugestie Bragga dały początek tzw. strukturalnej teorii tarcia smarów stałych.
- Wielu autorów uważa (41), (45), (46), że takie wyjaśnienia są nadmiernym uproszczeniem zagadnienia. Co prawda przyjmuje się, że energia wiązania atomów węgla znajdujących się w sąsiednich warstwach w graficie wynosi 4,18 do 83,72 J/mol (l do 20 kcal/mol), a pomiędzy atomami węgla ułożonymi w warstwie wynosi około 335 J/mol (80 kcal/mol), to jednak energia wiązań międzywarstwowych jest wielkością addytywną i suma energii tych wiązań może osiągać znaczne wartości. Wspomniani autorzy zasadniczą rolę w łatwym poślizgu między warstwami upatrują głównie w defektach struktury. Jako argument przeciw takiemu wyjaśnieniu własności smarujących materiałów o budowie warstwowej przedstawia się fakt, że kilka takich materiałów, jak np. mika, talk, dwusiarczek tytanu, pomimo że mają typową warstwową budowę, charakteryzują się wysokimi wartościami współczynnika tarcia. Dodatkowym argumentem jest fakt, że grafit oraz azotek boru w atmosferze powietrza, stanowiącej środowisko tarcia, wykazują małe wartości współczynników tarcia, podczas gdy w próżni wartość współczynnika tarcia ulega znacznemu zwiększeniu (35). W przypadku niektórych substancji występuje odwrotne zjawisko, tj. zmniejszenie wartości współczynnika tarcia w próżni (rys. 4.28, tabl. 4.12).
Rys. 4.28. Współczynnik tarcia halogenków kadmu w powietrzu i w próżni - Na podstawie powyższych rozważań można przypuszczać, że w wielu przypadkach dużą rolę w procesach tarcia smarów stałych odgrywają zjawiska sorpcyjne, bowiem w środowisku powietrza przebiega sorpcja gazów i pary wodnej na powierzchniach smarów stałych, natomiast w próżni tego rodzaju procesy nie zachodzą. Pod uwagę należy brać również procesy utleniania.
- Omówione zjawiska dały podstawę do zaproponowania przyjęcia tzw. adsorpcyjnej teorii tarcia smarów stałych. W teorii tej nie neguje się wpływu czynników strukturalnych, niemniej jednak zasadniczą rolę przypisuje się procesom adsorpcji i utleniania.
- R. H. Savage (48) stwierdził, że w przypadku grafitu występują zmiany wartości współczynnika tarcia w powietrzu, tlenie i próżni. Zaproponował on teoretyczne wyjaśnienie tego zjawiska. Stwierdził on ponadto, że współczynnik tarcia grafitu zostaje wydatnie obniżony (w porównaniu z wartością w próżni) przy kontakcie kryształów grafitu z łatwo kondensującymi się parami, jak np. amoniaku, benzenu czy n-heksanu, natomiast efektu takiego nie stwierdza się przy kontakcie z atmosferą azotu czy wodoru. Savage uważa, że aby grafit mógł spełniać rolę smaru stałego, musi nastąpić zmniejszenie energii powierzchniowej w wyniku adsorpcji. Zwłaszcza ważne jest zmniejszenie dużej energii powierzchniowej krawędzi i naroży kryształów, których addytywne oddziaływanie powoduje wystąpienie dużej wartości współczynnika tarcia oraz silnych procesów zużycia. Nieaktywność wodoru tłumaczy Savage małą zdolnością tego gazu do pokrywania powierzchni grafitu (dla wodoru 1 ¸ 4%, a dla tlenu 20 - 25% powierzchni). Mała aktywność została stwierdzona również w przypadku azotu.
Tablica 4.12.
Wartości współczynników tarcia niektórych smarów stałych
w próżni i w powietrzu (47)Ciało stałe Powietrze Próżnia Ciśnienie, µPa Grafit naturalny 0,19 0,44 0,798 Grafit pirolityczny 0,18 0,50 0,266 - BN
0,25 0,70 0,266 - MoSz
0,18 0,07 0,266 - WSz
0,17 0,13 0,399 - BiJ3
0,34 0,39 66,50 - LiOH
0,37 0,21 0,266 - NiJ2
0,48 0,44 26,60 - CdCl2
0,35 0,16 0,266 - CdJ2
0,24 0,18 0,266 - CdBr2
0,22 0,15 0,266 - SnS2
0,40 1,0 0,266 Ftalocjanina 0,35 0,33 133,30 - Podobne rezultaty otrzymał w swoich badaniach Rowe (35). Badał on zachowanie się grafitu w wysokiej próżni, w atmosferze tlenu, pary wodnej i innych łatwo kondensujących się par. Stwierdził, że wrażliwość grafitu na sorpcję pary wodnej w dużym stopniu zależy od rodzaju zastosowanego grafitu.
- Do ciekawych rezultatów doszli w swoich pracach Campbell i Kozak (50). Badali oni tarcie pręta grafitowego o obracającą się tarczę miedzianą. Stwierdzili, że bardzo dużą rolę odgrywa chropowatość i twardość tarczy. W przypadku polerowanej tarczy miedzianej bardzo małe zużycie grafitu występuje nawet w atmosferze suchego azotu. Podobny efekt uzyskuje się, gdy kryształy w pręcie grafitowym są zorientowane równolegle do powierzchni tarczy. W przypadku zwiększenia chropowatości powierzchni tarczy i przy prostopadłej do powierzchni tarcia orientacji kryształów grafitu, małe zużycie uzyskuje się jedynie w obecności tlenu, pary wodnej i innych łatwo kondensujących się par.
- Cannon (51) stwierdza, że dużą rolę odgrywa długość łańcuchów zaadsorbowanych na powierzchni cząstek sorbatu. Badano również wpływ temperatury na wartość współczynnika tarcia grafitu. Stwierdzono, że powyżej 50°C wartość jego wzrasta od 0,1 do 0,4. Wzrost ten przypisuje się desorpcji pewnej ilości pary wodnej oraz procesom utleniania powierzchniowego.
- Zwolennicy adsorpcyjnej teorii tarcia smarów stałych zwracają ponadto uwagę na procesy adhezyjne. Twierdzą oni, że przy tarciu smarów stałych znacznie ważniejszą cechą, niż budowa warstwowa smaru, jest jego zdolność do adhezji, na powierzchni smarowanego metalu. Dobre smarowanie uzyskuje się wówczas, gdy związanie adhezyjne z powierzchnią metalu jest silniejsze niż siły wiązań pomiędzy warstwami smaru stałego, co umożliwia poślizg. Należy zaznaczyć, że obecnie zarówno teoria strukturalna, jak i adsorpcyjna, nie są w stanie wyjaśnić wszystkich zaobserwowanych zjawisk występujących przy tarciu smarów stałych.
4.2.3. Zastosowanie smarów stałych
Smary stałe są stosowane w tych przypadkach, gdy użycie innych rodzajów smarów jest niemożliwe lub niecelowe. Mogą być one stosowane przy bardzo dużych naciskach jednostkowych, przy których utrzymanie trwałej warstwy innych rodzajów smarów jest niemożliwe. Smary stałe odznaczają się również bardzo dużą odpornością na wysokie temperatury, przy których inne rodzaje smarów ulegają rozkładowi cieplnemu. Swoje własności smarne zachowują one nawet w niskich temperaturach, dlatego też znalazły one zastosowanie w wielu urządzeniach kriogenicznych. Ze względu na to, że są ciałami stałymi, mogą one być stosowane do procesów smarowania w wysokiej próżni, w której inne rodzaje smarów ulegają odparowaniu i nie mogą spełniać swoich funkcji smarnych. Zazwyczaj są one odporne na silne oddziaływanie radiacyjne, co pozwala na wykorzystanie ich w technice jądrowej, natomiast duża odporność na oddziaływanie agresywnego środowiska (kwasów, zasad i innych agresywnych chemikalii) umożliwia ich stosowanie w szerokim zakresie do smarowania różnego rodzaju elementów i urządzeń w przemyśle chemicznym.- Duża trwałość smarów stałych pozwala na stosowanie ich w tych przypadkach, gdy jest konieczne jednorazowe nasmarowanie współpracujących elementów na cały okres pracy maszyny. Cecha ta ma również duże znaczenie przy trudnym lub niemożliwym w czasie eksploatacji dostępie do smarowanego skojarzenia trącego. Spośród wielu substancji stałych, odznaczających się zdolnością do zmniejszania wartości współczynnika tarcia, jako smary stałe mogą być stosowane tylko te substancje, które nie są higroskopijne czy rozpuszczalne w wodzie, jak również nie odznaczają się korozyjnością w stosunku do tworzyw, z których są wykonane smarowane elementy.
4.2.3.1. Grafit i dwusiarczek molibdenu
Najczęściej stosowanymi smarami stałymi są grafit i dwusiarczek molibdenu. Do smarowania w szczególnie wysokich temperaturach coraz częściej stosuje się również dwusiarczek wolframu. Grafit i dwusiarczek molibdenu (MoS2) są ciałami krystalicznymi (budowa warstwowa, heksagonalna, kolor szaro-czarny z metalicznym połyskiem). Twardość wg skali Mohsa grafitu wynosi 0,5 ¸ 1,5, dwusiarczku molibdenu-1-2. Gęstość grafitu wynosi 1,98 ¸ 2,38 kg/m³, MoS2 - 4,7 ¸ 4,8 kg/m³. Grafit jest termicznie trwały w próżni do temperatury 3500°C, w atmosferze powietrza do 550°C. W atmosferze tlenu utlenia się podobnie jak MoS2już w temperaturze pokojowej. Dwusiarczek molibdenu odznacza się mniejszą odpornością cieplną niż grafit (w próżni do 1200°C, w argonie do 1343°C, w atmosferze powietrza do 450°C). Utlenianie jest procesem działającym destrukcyjnie na grafit i dwusiarczek molibdenu. Szybkość utleniania zależy od wartości temperatury, w której przebiega proces, od stężenia tlenu, od czasu oddziaływania tlenu oraz od powierzchni kontaktu, a więc od wielkości ziarn kryształów. Dwusiarczek molibdenu w wyniku utleniania przechodzi w trójtlenek molibdenu (MoO3), który nie ma własności smaru stałego i pogarsza proces smarowania. Obszerne badania intensywności utleniania MoS2 w różnych temperaturach przeprowadził Vineal (56) (tabl. 4.13).Tablica 4.13.
Intensywność utlenienia dwusiarczku molibdenu w różnych temperaturach (wg Vineala)Temperatura °C Czas ogrzewania, h 1 3 6 18 24 48 72 96 zawartość procentowa trójtlenku molibdenu 250 < 0,1 < 0,1 < 0,1 < 0,1 0,2 0,4 0,5 0,6 300 1,0 3,2 5,0 15,0 19,5 24,3 26,0 27,3 400 2,9 12,1 16,0 22,5 24,5 27,9 27,0 46,0 500 57,2 74,5 79,3 80,2 84,7 94,7 96,0 96,5 - Obydwa podstawowe smary stałe mogą być otrzymywane ze źródeł naturalnych (ze złóż kopalnych - kopalny grafit i molibdenit) lub syntetycznie. Grafit syntetyczny otrzymuje się przez uwęglanie i grafityzację w wysokich temperaturach substancji bogatych w węgiel (np. węglowodorów), a syntetyczny dwusiarczek molibdenu przez zastosowanie wielu różnych metod, jak np. :
- z pierwiastków Mo+2S ® MoS2, przez prażenie w rurze żelaznej,
- z trójtlenku molibdenu w obecności siarkowodoru
MoO3 + 3H2S ® MoS2 + 3H2O + S - z molibdenianu amonu
(NH4)2MoO4 + 2H2S ® (NH4)2MoS2 + 4H2O
(NH4)2MoS2+2H2 ® MoS2 MoS2 + H2S + (NH4)2S - z molibdenianu amonu przez stapianie z siarką i za pomocą wielu innych metod.
- W Polsce produkcję syntetycznego dwusiarczku molibdenu opracowali Maciaszek (58) i Haś (57).
- Oddzielnym zagadnieniem jest sposób nanoszenia wspomnianych smarów stałych na powierzchnie smarowane. Smary te mogą być nanoszone lub kontaktowane ze smarowanymi powierzchniami w postaci zwartej (brykiety, ołówki i szczotki smarujące) lub w postaci rozdrobnionej, tj. w postaci proszku. Proszki mogą być wcierane w smarowane powierzchnie lub też nanoszone w postaci zawiesin i aerosoli. Stosuje się zawiesiny wodne smarów stałych lub w łatwo lotnych rozpuszczalnikach. Sporządzane są również różnego rodzaju pasty. Stosunkowo duże ilości smarów stałych są zawieszane w olejach smarnych oraz w smarach plastycznych. Dla zwiększenia przyczepności do podłoża sproszkowane smary stałe są zawieszane w lakierach, którymi maluje się smarowane powierzchnie, lub też w szkle wodnym (roztwór krzemianu sodu), które powodują powiązanie z podłożem. W szerokim zakresie, jako materiały wiążące, wykorzystywane są różnego rodzaju żywice syntetyczne. Grafit i dwusiarczek molibdenu wprowadza się również do łożysk ze spieków oraz do plastycznych tworzyw niskotarciowych.
4.2.3.2. Miękkie metale
Miękkie metale znalazły zastosowanie w trybotechnice w postaci sproszkowanej, jak również w postaci zwartej, jako niskotarciowe materiały łożyskowe. Największe zastosowanie w postaci sproszkowanej znalazły proszki miedzi, glinu, cyny, ołowiu, a w niektórych przypadkach i srebra. Stosowane są również proszki stopów miękkich metali.- Miękkie metale są ponadto nanoszone w postaci bardzo cienkich powłok (grubość ok. 10 -6 µm na twardym podłożu, w celu zabezpieczenia tego podłoża przed zatarciami w przypadku przerwania smarnej warstwy olejowej. Stanowią więc one dodatkowe zabezpieczenie elementów przed zatarciem. Do wytwarzania miękkich powłok na twardym podłożu najczęściej są stosowane: ind, ołów, gal i kadm. Szczególnie dobre rezultaty otrzymuje się w przypadku powłok z indu.
4.2.3.3. Polimery niskotarciowe
Spośród polimerów odznaczających się małą wartością współczynnika tarcia największe zastosowanie znalazły: teflon (policzterofluoroetylen) oraz poliamidy (nylon).- Teflon, jako polimer niskotarciowy, ma zarówno wiele zalet, jak i wad. Z zalet należy wymienić: najmniejszą wartość współczynnika tarcia z dotychczas znanych materiałów (bez smarowania), brak zjawiska „stick-slip" podczas tarcia, obojętność chemiczna, niewrażliwość na działanie agresywnego środowiska oraz możliwość pracy w stosunkowo wysokich temperaturach (do 300°C). Do wad, w poważnym stopniu ograniczających jego zastosowania, należą: mała przewodność cieplna, duża rozszerzalność termiczna, „płynięcie na zimno" pod wpływem obciążeń ściskających oraz mała odporność na zużycie. Dla wyeliminowania wad teflonu stosuje się go w postaci wzmocnionej włóknem szklanym, metalowym, azbestowym lub innym oraz tworzy się mieszaniny z polimerami o dużej wytrzymałości mechanicznej, często z zastosowaniem wypełniacza w postaci proszku grafitowego czy też MoS2. Praktykowane jest również nanoszenie cienkich powłok z teflonu na metalowe podłoża lub też wypełnianie materiałów porowatych teflonem (np. łożyska wg patentu firmy Glacier). Teflon w postaci sproszkowanej może być również nanoszony na powierzchnie smarowane w postaci zawiesin (np. past, w smarach plastycznych itp.). Ten sposób wykorzystania teflonu w technice smarowniczej zapewnia uzyskanie bardzo dobrych efektów.
- Sproszkowane poliamidy, jako materiały niskotarciowe, nie zapewniają takich efektów jak teflon, ale w postaci zwartej są w szerokim zakresie stosowane jako materiał na elementy ślizgowe łożysk pracujących w umiarkowanych temperaturach i przy niewysokich obciążeniach. W szerokim zakresie są również stosowane poliamidy z wypełniaczem grafitowym lub z MoS2.
- Wielu autorów uważa (41), (45), (46), że takie wyjaśnienia są nadmiernym uproszczeniem zagadnienia. Co prawda przyjmuje się, że energia wiązania atomów węgla znajdujących się w sąsiednich warstwach w graficie wynosi 4,18 do 83,72 J/mol (l do 20 kcal/mol), a pomiędzy atomami węgla ułożonymi w warstwie wynosi około 335 J/mol (80 kcal/mol), to jednak energia wiązań międzywarstwowych jest wielkością addytywną i suma energii tych wiązań może osiągać znaczne wartości. Wspomniani autorzy zasadniczą rolę w łatwym poślizgu między warstwami upatrują głównie w defektach struktury. Jako argument przeciw takiemu wyjaśnieniu własności smarujących materiałów o budowie warstwowej przedstawia się fakt, że kilka takich materiałów, jak np. mika, talk, dwusiarczek tytanu, pomimo że mają typową warstwową budowę, charakteryzują się wysokimi wartościami współczynnika tarcia. Dodatkowym argumentem jest fakt, że grafit oraz azotek boru w atmosferze powietrza, stanowiącej środowisko tarcia, wykazują małe wartości współczynników tarcia, podczas gdy w próżni wartość współczynnika tarcia ulega znacznemu zwiększeniu (35). W przypadku niektórych substancji występuje odwrotne zjawisko, tj. zmniejszenie wartości współczynnika tarcia w próżni (rys. 4.28, tabl. 4.12).
